Síndrome Nefrótico y Linfoma de Hodgkin. Reporte de dos casos.
Rev Med Hered v.22 n.4 Lima oct./dic. 2011
NTRODUCCIÓN
El síndrome nefrótico (SN) es un trastorno que se caracteriza por proteinuria masiva y persistente, hipercolesterolemia, hipoalbuminemia y otras manifestaciones secundarias a la proteinuria masiva, como consecuencia de una lesión que altera la barrera de filtración glomerular.
Puede ser primaria o secundaria. Las primarias son producidas por una variedad de glomerulonefritis (GN) en las cuales no se conoce la causa de la enfermedad, siendo las más frecuentes la enfermedad de cambios mínimos (ECM), la glomeruloesclerosis focal y segmentaria (GEFS) y la GN membranoproliferativa (GNMP) (1,2,3). En las secundarias, el compromiso renal ocurre como parte de una enfermedad sistémica como: colagenopatías (Lupus eritematoso sistémico, Poliarteritis nodosa, etc.), infecciones (VIH, hepatitis B y C, paludismo, etc.), intoxicaciones (bismuto, mercuriales, sales de oro, etc.) y algunas veces en neoplasias: tumor de Wilms, neuroblastoma, leucemia y linfoma tanto Hodgkin como no Hodgkin (4).
Son pocos los reportes en pediatría sobre la relación entre el síndrome nefrótico (SN) y los tumores malignos, habiéndose descrito por primera vez en 1922 (5,6). Hoy se sabe que en las neoplasias sólidas como los carcinomas, el SN suele corresponder, desde el punto de vista anatomopatológico, a una glomerulonefritis membranosa (GM), mientras que en las neoplasias hematológicas, el patrón histopatológico que predomina es la ECM, asociada con mayor frecuencia al linfoma de Hodgkin; en el linfoma no Hodgkin las lesiones renales son diversas y complejas (7,8). En vista de la poca información sobre el tema, reportamos dos casos de síndrome nefrótico asociado a linfoma Hodgkin en niños.
Caso clínico 1
Varón de 7 años, con historia de 9 meses de edema generalizado. Al ingreso tenía presión arterial: 110/60 mm Hg y anasarca. Los exámenes de laboratorio mostraron: Hematocrito 37%, plaquetas 700 000, urea 15,5 mg/dl, creatinina 0,4mg/dl, colesterol 500 mg/dl, triglicéridos 370 mg/dl; proteinuria: 53,3mg/m2/h, y depuración de creatinina en 146 ml/min x 1,73m2. El examen de orina mostró proteinuria 500 mg/dl y el sedimento era normal. Con el diagnóstico de síndrome nefrótico, recibió terapia con prednisona 60 mg/m2/d por 3 meses con buena respuesta.
Cuatro meses después fue hospitalizado por presentar celulitis en el lado derecho de la cara. En dicha hospitalización se encontró una masa mediastinal en la radiografía de tórax; ante la sospecha de TBC ganglionar se inició tratamiento específico, recibiéndolo hasta la fase dos del esquema 1.
Un mes después presentó recaída del síndrome nefrótico, lo que motivó su hospitalización, iniciando corticoterapia. Al examen clínico se le encontró un ganglio supraclavicular, realizándose la biopsia ganglionar, siendo el informe Linfoma de Hodgkin clásico. Luego de la quimioterapia hubo remisión de la enfermedad hematológica y de la enfermedad renal, esta última luego de un año de tratamiento.
Caso clínico 2
Varón de 5 años con historia de un mes de edema palpebral y orinas espumosas. Al examen tenía presión arterial 110/70 mm Hg. Los exámenes de laboratorio mostraron: Hematocrito 32% plaquetas 307 000, VSG 52 mm/hora, urea 24 mg/dl, creatinina 0,7mg/dl, colesterol 227,4 mg/dl, C3 180 mg/dl, proteinuria 188 mg/m2/h. El examen de orina sólo mostró proteinuria (+); En la radiografía de tórax se observó un nódulo pulmonar solitario y el PPD era de 15mm. Además se encontró Arco Quinto negativo.
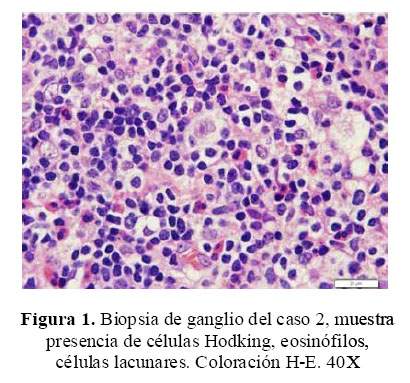
Se inició tratamiento antituberculoso (que se descontinuó luego de 20 días, posterior a la biopsia, permaneciendo con isoniacida), se realizó una biopsia pulmonar a "cielo abierto" y de un ganglio supraclavicular ( Figura 1) cuyo resultado fue linfoma de Hodgkin celularidad mixta. Recibió quimioterapia, remitiendo el cuadro hematológico. La enfermedad renal remitió a los 6 meses.
DISCUSIÓN
Los linfomas representan aproximadamente el 15% de las neoplasias pediátricas y se ubican en el tercer lugar en frecuencia luego de las leucemias y los tumores cerebrales malignos. En EEUU la frecuencia de linfoma es 3% en niños menores de 5 años y 24% en edades entre 15 y 19 años, mientras que en España la incidencia de linfoma es 15% (9).
En un estudio realizado en Cuzco, se encontró que el linfoma de Hodgkin representó el 6,7% de las neoplasias hematológicas (10). Los linfomas pueden ser de dos tipos: No Hodgkin y Hodgkin. El linfoma no Hodgkin es más frecuente en los niños más pequeños mientras que el linfoma Hodgkin lo es en adolescentes. El linfoma de Hodgkin representa el 30-40% de todos los linfomas (11); se caracteriza por el aumento de volumen de los ganglios linfáticos supraclavicular o cervical, usualmente poco dolorosos y de consistencia firme, encontrándose en el 60% compromiso intratorácico (masa mediastinal). Además pueden presentar fiebre, pérdida de peso, sudoración nocturna, prurito, y otras como, dolor abdominal, ictericia, edema periférico, dolor óseo, signos de compresión medular, obstrucción ureteral y síndrome nefrótico.
Los tumores pueden causar síndromes paraneoplásicos, estos son raros y pueden ser el primer signo o la sintomatología principal de la enfermedad.
Se ha reportado hematuria o proteinuria en pacientes con tumores y en necropsias, se ha encontrado 17-30% de pacientes con neoplasias con cambios glomerulares menores. Los reportes en pediatría son raros (12). La proteinuria puede ser detectada en pacientes con cáncer, sin embargo, el síndrome nefrótico es raro. El SN puede preceder, coincidir o seguir al diagnóstico de la enfermedad neoplásica. Richmond et al, estudiaron 696 pacientes con linfoma y médula ósea y encontraron que el compromiso renal en la enfermedad de Hodgkin era 13% (13). La incidencia de SN en linfoma de Hodgkin es 0,4% y suele coincidir con el diagnóstico o precederlo por meses o años; en ocasiones es el síntoma debut o se asocia a las recaídas, ya que la corticoterapia puede enmascarar la aparición florida del mismo (14,15).
En tumores sólidos, el SN es con frecuencia secundario, siendo el patrón histopatológico de una glomerulonefritis membranosa, mientras que en neoplasias hematológicas, el patrón predominante suele corresponder a enfermedad de cambios mínimos; pero se ha reportado otras alteraciones renales, como e la glomeruloesclerosis focal y segmentaria (GEFS) que siendo corticoresistente, remite con quimioterapia (16).
Cuando el SN se asocia a la enfermedad de Hodgkin, la lesión más común suele ser la de cambios mínimos, seguida de GEFS, mesangioproliferativa, membranosa, membranoproliferativa o por depósito de complejos inmunes. Los pacientes con linfoma de Hodgkin variedad celularidad mixta son más propensos a desarrollar nefropatía por cambios mínimos (9,17).
Cuando un SN es dependiente o resistente a corticoides debe sospecharse de enfermedad de Hodgkin concomitante (18).
Con relación a la fisiopatología del SN en el curso de un linfoma, algunos autores mencionan ciertos factores de riesgo como: Historia familiar de enfermedad de Hodgkin, infección por Ebstein Barr, estado socioeconómico, contactos sociales, trasplante renal y cardiaco (19). Los mecanismos por los que se produce un SN en un paciente con linfoma son especulativos, sin embargo se ha intentado explicar por desórdenes de los linfocitos T, vía procesos mediados por linfoquinas, por depósito de complejos inmunes, compuestos en parte por antígenos asociados al tumor, que causan depósitos subepiteliales en el glomérulo, por coagulación intravascular o amiloidosis. Así la función de las células T alteradas se asocia a: permeabilidad glomerular alterada por citoquinas, glomeruloesclerosis, sobreexpresión de pronto-oncogenes (20).
Ninguno de los dos casos tuvo biopsia renal, sin embargo al iniciar tratamiento por la enfermedad de fondo mejoró el cuadro renal, lo cual apoya la asociación con enfermedad de cambios mínimos. En los últimos controles realizados, ambos pacientes sino tenían evidencia de alteración renal.
Por todo esto en aquellos niños que cursan con síndrome nefrótico con episodios de recaídas, corticoresistencia, corticodependencia, y masas se debe sospechar no sólo en procesos infecciosos sino también en alguna enfermedad linfoproliferativa; de allí la importancia de reportar estos dos casos


