Anemia hemolitica autoinmune
Mostrando entradas con la etiqueta anemias hemoliticas. Mostrar todas las entradas
Mostrando entradas con la etiqueta anemias hemoliticas. Mostrar todas las entradas
miércoles, 19 de diciembre de 2018
Hemoglobinura paroxística nocturna: terapia con Eculizumab
Hemoglobinura paroxística nocturna: terapia con Eculizumab
El inhibidor del complemento eculizumab en la hemoglobinuria paroxística nocturna.Autor: Peter Hillmen, M.B., Ch.B., Ph.D., Neal S. Young, M.D., Jörg Schubert, M.D Fuente: NEJM: Volume 355:1233-1243 September 21, 2006 Number 12 The Complement Inhibitor Eculizumab in Paroxysmal Nocturnal Hemoglobinuria
Antecedentes
Analizamos la seguridad y la eficacia del eculizumab, un anticuerpo monoclonal humanizado contra la proteína terminal del complemento C5, que inhibe la activación final del complemento, en los pacientes con hemoglobinuria paroxística nocturna (HPN).
Analizamos la seguridad y la eficacia del eculizumab, un anticuerpo monoclonal humanizado contra la proteína terminal del complemento C5, que inhibe la activación final del complemento, en los pacientes con hemoglobinuria paroxística nocturna (HPN).
Métodos
Llevamos a cabo un ensayo de fase 3 multicéntrico, doble ciego, aleatorizado y controlado con placebo. Los pacientes recibieron un placebo o eculizumab por vía intravenosa; las dosis de eculizumab fueron de 600 mg semanales durante 4 semanas, una dosis de 900 mg una semana después y a continuación 900 mg cada dos semanas hasta la semana 26.
Los dos criterios principales de valoración fueron la estabilización de los niveles de hemoglobina y el número de unidades de concentrados de hematíes transfundidas. Se evaluaron también los indicadores bioquímicos de hemólisis intravascular y la calidad de vida de los pacientes.
Resultados
Se aleatorizó a 87 pacientes. Se alcanzó la estabilización de los niveles de hemoglobina sin transfusiones en el 49% de los pacientes (21 de 43) asignados al eculizumab y en ninguno (0 de 44) de los asignados al placebo (p<0,001).
Durante el estudio, se administró una mediana de 0 unidades de concentrados de hematíes en el grupo tratado con el eculizumab, en comparación con 10 unidades en el grupo que recibió el placebo (p<0,001).
El eculizumab redujo la hemólisis intravascular, como demuestra una mediana del área bajo la curva para el lactato-deshidrogenasa frente al tiempo (en días) un 85,8% más baja en el grupo tratado con eculizumab, en comparación con el grupo que recibió el placebo (58.587 frente a 411.822 U por litro; p<0,001).
Se observaron también mejorías clínicamente significativas en la calidad de vida, determinada mediante las puntuaciones en el instrumento para la evaluación terapéutica funcional de enfermedades crónicas sobre el cansancio (p<0,001) y el cuestionario sobre la calidad de la vida de la Organización europea para la investigación y el tratamiento del cáncer.
De los 87 pacientes, 4 en el grupo tratado con eculizumab y 9 en el grupo que recibió el placebo presentaron acontecimientos adversos serios, ninguno de los cuales se consideró relacionado con el tratamiento; todos estos pacientes se recuperaron sin secuelas.
Se aleatorizó a 87 pacientes. Se alcanzó la estabilización de los niveles de hemoglobina sin transfusiones en el 49% de los pacientes (21 de 43) asignados al eculizumab y en ninguno (0 de 44) de los asignados al placebo (p<0,001).
Durante el estudio, se administró una mediana de 0 unidades de concentrados de hematíes en el grupo tratado con el eculizumab, en comparación con 10 unidades en el grupo que recibió el placebo (p<0,001).
El eculizumab redujo la hemólisis intravascular, como demuestra una mediana del área bajo la curva para el lactato-deshidrogenasa frente al tiempo (en días) un 85,8% más baja en el grupo tratado con eculizumab, en comparación con el grupo que recibió el placebo (58.587 frente a 411.822 U por litro; p<0,001).
Se observaron también mejorías clínicamente significativas en la calidad de vida, determinada mediante las puntuaciones en el instrumento para la evaluación terapéutica funcional de enfermedades crónicas sobre el cansancio (p<0,001) y el cuestionario sobre la calidad de la vida de la Organización europea para la investigación y el tratamiento del cáncer.
De los 87 pacientes, 4 en el grupo tratado con eculizumab y 9 en el grupo que recibió el placebo presentaron acontecimientos adversos serios, ninguno de los cuales se consideró relacionado con el tratamiento; todos estos pacientes se recuperaron sin secuelas.



Conclusiones El eculizumab es un tratamiento eficaz para la HPN.
* Acceda al texto completo en inglés haciendo click aquí
(Número en ClinicalTrials.gov: NCT00122330 [ClinicalTrials.gov] .)
Fuente de información:
Complicaciones a largo plazo del síndrome urémico hemolítico
Complicaciones a largo plazo del síndrome urémico hemolítico
Las características clínicas clásicas del SUH incluyen la tríada de anemia hemolítica microangiopática, trombocitopenia e insuficiencia renal aguda.Autor: Dra. Sharon P. Andreoli* Fuente: Department of Pediatrics and the Herman B. Wells Center for Pediatric Research. Indiana University School of Medicine, Indianapolis, IN, USA. Arch.Latin.Nefr.Ped. 2003; 3(1)
Desarrollo
El síndrome urémico hemolítico (SUH) es una causa frecuente de insuficiencia renal aguda en niños y provoca morbilidad y mortalidad significativas durante la fase aguda, así como complicaciones a largo plazo. Las características clínicas clásicas del SUH incluyen la tríada de anemia hemolítica microangiopática, trombocitopenia e insuficiencia renal aguda.(1-4) No obstante, los estudios epidemiológicos en brotes epidémicos de SUH con colitis hemorrágica y diarrea demostraron claramente que algunos pacientes presentan anemia hemolítica o trombocitopenia con escasas evidenciasde compromiso renal, mientras queotros presentan importante enfermedad renal con un recuento plaquetario normal o hemólisis mínima.(1-4)
El SUH se clasifica como la variante epidémica, con diarrea (típico) o como SUH esporádico, sin diarrea (atípico). El SUH atípico puede ser familiar y asociarse con mutaciones en el factor H o con medicamentos como ciclosporina, píldoras anticonceptivas o mitomicina.(5,6) Este trabajo describira las manifestaciones clínicas del SUH típico con diarrea durante la fase aguda, sus complicaciones a largo plazo y los estudios recientes sobre la fisiopatología del SUH asociado con STEC, con énfasis en el papel de la respuesta inflamatoria en la fisiopatología de la enfermedad.
Epidemiología de las infecciones por E. Coli productora de Shiga Toxina
Las cepas de E. coli productoras de shiga toxina (STEC) se relacionaron por primera vez con la enfermedad en seres humanos a comienzos de la década de 1980.(1-3) Varios brotes de colitis hemorrágica y SUH, ampliamente publicitados, resaltaron la morbilidad y mortalidad de la infección con E. coli productora de verocitotoxina. En 1993, una epidemia en el Oeste de EE.UU. produjo varios casos de SUH grave con considerables complicaciones extrarrenales.(7)
En 1996, más de 5.000 escolares japoneses se infectaron con E. coli productora de verocitotoxina; una epidemia en Escocia produjo 20 muertes; en otra epidemia de EE.UU., la fuente de transmisión había sido un jugo de manzana contaminado. Varios millones de libras de hamburguesas contaminadas fuerondescartadas en EE.UU. En estudios caso-control, el consumo de hamburguesas de carne bovina o de carne fría cortada en rebanadas se asoció con infección por E. coli 0157:H7.(10)
Aunque las hamburguesas poco cocidas son el vector más frecuente de la infección por O157:H7, el jugo de manzana, los brotes de rábano, las salchichas y otros alimentos han sido implicados en la propagación de infecciones por STEC. En EE.UU. y Europa, el serotipoO157:H7 es el implicado con mayor frecuencia en las infecciones con producción de VT, pero en otras áreas del mundo se están informando como patógenos importantes cepas no 0157:H7.(11-13) La E. coli O111:H- produjo una gran epidemia de colitis hemorrágica y SUH en Australia y otros sero tipos también se han asociado con colitis hemorrágica y SUH. (11-13)
La tasa de progresión a SUH tras la infección con STEC es de 5 a 15%, aproximadamente. En niños menores de 5 años, la incidencia de SUH es de 12,9% en comparación con un riesgo de 6,8% y 8% en los niños de 5 a 9,9 años y mayores de 10 años, respectivamente.(4) En otro estudio, los niños con recuentos leucocitarios mayores de 13.000/mm3 durante los primeros 3 días de la infección causada por STEC, tuvieron un riesgo de desarrollar SUH siete veces mayor que pacientes de edades comparables con un recuento leucocitario menor de 13.000/mm3.
La administración de agentes depresores de la motilidad gastrointestinal también se asoció con mayor riesgo de SUH.(14) Un estudio reciente demostró que los niños con colitis hemorrágica asociada con STEC que reciben tratamiento con antibióticos, tienen muchas más probabilidades de progresara SUH. Por el momento no se conocen los factores ambientales o genéticos que podrían predisponer a la progresión de la colitis hemorrágica asociada con STEC a SUH. Se sugirió que las alteraciones en el gen del Factor H, descriptas recientemente en pacientes con SUH atípico, también podrían ser importantes en el SUH epidémico con diarrea.(5)Manifestaciones clínicas y complicaciones a largo plazo
El SUH se clasifica como la variante epidémica, con diarrea (típico) o como SUH esporádico, sin diarrea (atípico). El SUH atípico puede ser familiar y asociarse con mutaciones en el factor H o con medicamentos como ciclosporina, píldoras anticonceptivas o mitomicina.(5,6) Este trabajo describira las manifestaciones clínicas del SUH típico con diarrea durante la fase aguda, sus complicaciones a largo plazo y los estudios recientes sobre la fisiopatología del SUH asociado con STEC, con énfasis en el papel de la respuesta inflamatoria en la fisiopatología de la enfermedad.
Epidemiología de las infecciones por E. Coli productora de Shiga Toxina
Las cepas de E. coli productoras de shiga toxina (STEC) se relacionaron por primera vez con la enfermedad en seres humanos a comienzos de la década de 1980.(1-3) Varios brotes de colitis hemorrágica y SUH, ampliamente publicitados, resaltaron la morbilidad y mortalidad de la infección con E. coli productora de verocitotoxina. En 1993, una epidemia en el Oeste de EE.UU. produjo varios casos de SUH grave con considerables complicaciones extrarrenales.(7)
En 1996, más de 5.000 escolares japoneses se infectaron con E. coli productora de verocitotoxina; una epidemia en Escocia produjo 20 muertes; en otra epidemia de EE.UU., la fuente de transmisión había sido un jugo de manzana contaminado. Varios millones de libras de hamburguesas contaminadas fuerondescartadas en EE.UU. En estudios caso-control, el consumo de hamburguesas de carne bovina o de carne fría cortada en rebanadas se asoció con infección por E. coli 0157:H7.(10)
Aunque las hamburguesas poco cocidas son el vector más frecuente de la infección por O157:H7, el jugo de manzana, los brotes de rábano, las salchichas y otros alimentos han sido implicados en la propagación de infecciones por STEC. En EE.UU. y Europa, el serotipoO157:H7 es el implicado con mayor frecuencia en las infecciones con producción de VT, pero en otras áreas del mundo se están informando como patógenos importantes cepas no 0157:H7.(11-13) La E. coli O111:H- produjo una gran epidemia de colitis hemorrágica y SUH en Australia y otros sero tipos también se han asociado con colitis hemorrágica y SUH. (11-13)
La tasa de progresión a SUH tras la infección con STEC es de 5 a 15%, aproximadamente. En niños menores de 5 años, la incidencia de SUH es de 12,9% en comparación con un riesgo de 6,8% y 8% en los niños de 5 a 9,9 años y mayores de 10 años, respectivamente.(4) En otro estudio, los niños con recuentos leucocitarios mayores de 13.000/mm3 durante los primeros 3 días de la infección causada por STEC, tuvieron un riesgo de desarrollar SUH siete veces mayor que pacientes de edades comparables con un recuento leucocitario menor de 13.000/mm3.
La administración de agentes depresores de la motilidad gastrointestinal también se asoció con mayor riesgo de SUH.(14) Un estudio reciente demostró que los niños con colitis hemorrágica asociada con STEC que reciben tratamiento con antibióticos, tienen muchas más probabilidades de progresara SUH. Por el momento no se conocen los factores ambientales o genéticos que podrían predisponer a la progresión de la colitis hemorrágica asociada con STEC a SUH. Se sugirió que las alteraciones en el gen del Factor H, descriptas recientemente en pacientes con SUH atípico, también podrían ser importantes en el SUH epidémico con diarrea.(5)Manifestaciones clínicas y complicaciones a largo plazo
Las principales manifestaciones clínicas del SUH incluyen la tríada de trombocitopenia, anemia hemolítica e insuficiencia renal aguda secundariamente a una colitis hemorrágica. Aunque el riñón y el aparato gastrointestinal son los órganos afectados con mayor frecuencia en el SUH, también pueden producirse evidencias de compromiso del sistema nervioso central, páncreas, esqueleto y miocardio.(7,15,16)
Durante la fase aguda del SUH, las biopsias de riñón revelan una lesión microvascular caracterizada por depósitos de microtrombos junto con células endoteliales glomerulares tumefactas y desprendidas e infiltración de células inflamatorias.(17,18) Se describieron cambios patológicos similares en otros órganos, incluidos colon, SNC, páncreas, músculo esquelético y miocárdico.
Estos hallazgos demuestran que el SUH es una enfermedad sistémica caracterizada por lesión de las células endoteliales. Se desconoce el mecanismo de esta lesión microvascular pero las evidencias sugieren la intervención de la VT, con cambios en el perfil anticoagulante normal de la célula endotelial hacia un estado procoagulante. Los avances técnicos en el tratamiento de diálisis y la mejor atención de los niños en estado crítico permitieron una disminución significativa de la mortalidad aguda por SUH, por lo que son más evidentes las complicaciones crónicas en los sobrevivientes a largo plazo.
Algunos niños nunca recuperan la función renal y requieren tratamiento sustitutivo prolongado, aunque los que se recuperan tienen riesgo de desarrollo tardío de nefropatía.(19-20) Además, algunos niños presentan otros problemas extrarrenales, incluidos defectos neurológicos, diabetes mellitus insulinodependiente, insuficiencia pancreática y complicaciones gastrointestinales.(21-23) Por lo tanto, el SUH es una enfermedad con mortalidad y morbilidad multisistémicas sustanciales.
Varios estudios demostraron que los niños que se recuperan del episodio agudo de SUH tienen riesgo de presentar complicaciones alargo plazo, como hipertensión, insuficiencia renal, insuficiencia renal terminal y diabetes mellitus insulino dependiente. Seigler observó que 39% de 61 niños con antecedentes de SUH presentaron complicaciones tardías, como hipertensión, proteinuria e insuficiencia renal durante una media de 9,6 años después del episodio agudo.(24)
La duración de la oligoanuria fue el mejor indicador predictivo de complicaciones tardías. Caletti et al. también demostraron que el hallazgo histológico de esclerosis focal y segmentaria y hialinosis se observa varios años después del SUH. En ese estudio, sólo un 25% de los niños tuvieron función renal normal durante el seguimiento a largo plazo.(20) Gagnadouz et al., evaluaron a 29 niños de15 a 25 años después de la fase aguda del SUH.(25) Sólo 10 eran normales; 12 eran hipertensos, 3 sufrían insuficiencia renal crónica y 4 presentaban nefropatía terminal.
Se produjeron secuelas graves en niños con oligoanuria de7 días o más. La histología renal fue el mejor indicador predictivo de complicaciones a largo plazo. Moghdal y colaboradores realizaron biopsias renales en niños con antecedentes de SUH con proteinuria.(19) La mayoría de ellos presentaba esclerosis global y segmentaria con fibrosis intersticial. Los autores concluyeron que los niños con función renal normal y proteinuria leve presentan considerable daño renal y tienen riesgo de desarrollar insuficiencia renal tardíamente.(19)
Perlstein demostró que la carga oral de proteínas en 17 niños con antecedentes de SUH revelaba una reserva funcional renal disminuida en pacientes con antecedentes de SUH que tenían función renal y presión arterial normales, en comparación con niños sin tales antecedentes. Este estudio sugiere que la reserva funcional renal en niños con SUH puede estar disminuida a pesar de que la función renal y la presión se mantengan en valores normales.(26)
Los autores señalaron que no se conoce la importancia a largo plazo de este hallazgo y aún debe ser investigada, pero el estudio sugiere que la reserva funcional renal puede estar disminuida a pesar de la recuperación normal y que los niños con SUH requieren seguimiento prolongado. En el estudio de Spizzirri et al., 92,5% de los pacientes sin anuria se recuperaron, pero uno presentó proteinuria aislada, 2 tuvieron una disminución leve del índice de filtración glomerular (IFG) y uno sufrió insuficiencia renal terminal 19 años después de este episodio de SUH.(27) Además, 86% de los pacientes con proteinuria en el control de seguimiento del primer año tuvieron alteraciones renales al finalizar el seguimiento, pero un 25% de los pacientes sin proteinuria al año desarrollaron secuelas crónicas después de un lapso variable.
En resumen, los niños que aparentan haberse recuperado del SUH pueden presentar complicaciones tardías. Por lo tanto, es necesario determinar la fisiopatología de este síndrome para disminuir la mortalidad aguda y la morbilidad alargo plazo de esta enfermedad.
Mecanismos del daño ceñular mediado por la Shiga Toxina
Durante la fase aguda del SUH, las biopsias de riñón revelan una lesión microvascular caracterizada por depósitos de microtrombos junto con células endoteliales glomerulares tumefactas y desprendidas e infiltración de células inflamatorias.(17,18) Se describieron cambios patológicos similares en otros órganos, incluidos colon, SNC, páncreas, músculo esquelético y miocárdico.
Estos hallazgos demuestran que el SUH es una enfermedad sistémica caracterizada por lesión de las células endoteliales. Se desconoce el mecanismo de esta lesión microvascular pero las evidencias sugieren la intervención de la VT, con cambios en el perfil anticoagulante normal de la célula endotelial hacia un estado procoagulante. Los avances técnicos en el tratamiento de diálisis y la mejor atención de los niños en estado crítico permitieron una disminución significativa de la mortalidad aguda por SUH, por lo que son más evidentes las complicaciones crónicas en los sobrevivientes a largo plazo.
Algunos niños nunca recuperan la función renal y requieren tratamiento sustitutivo prolongado, aunque los que se recuperan tienen riesgo de desarrollo tardío de nefropatía.(19-20) Además, algunos niños presentan otros problemas extrarrenales, incluidos defectos neurológicos, diabetes mellitus insulinodependiente, insuficiencia pancreática y complicaciones gastrointestinales.(21-23) Por lo tanto, el SUH es una enfermedad con mortalidad y morbilidad multisistémicas sustanciales.
Varios estudios demostraron que los niños que se recuperan del episodio agudo de SUH tienen riesgo de presentar complicaciones alargo plazo, como hipertensión, insuficiencia renal, insuficiencia renal terminal y diabetes mellitus insulino dependiente. Seigler observó que 39% de 61 niños con antecedentes de SUH presentaron complicaciones tardías, como hipertensión, proteinuria e insuficiencia renal durante una media de 9,6 años después del episodio agudo.(24)
La duración de la oligoanuria fue el mejor indicador predictivo de complicaciones tardías. Caletti et al. también demostraron que el hallazgo histológico de esclerosis focal y segmentaria y hialinosis se observa varios años después del SUH. En ese estudio, sólo un 25% de los niños tuvieron función renal normal durante el seguimiento a largo plazo.(20) Gagnadouz et al., evaluaron a 29 niños de15 a 25 años después de la fase aguda del SUH.(25) Sólo 10 eran normales; 12 eran hipertensos, 3 sufrían insuficiencia renal crónica y 4 presentaban nefropatía terminal.
Se produjeron secuelas graves en niños con oligoanuria de7 días o más. La histología renal fue el mejor indicador predictivo de complicaciones a largo plazo. Moghdal y colaboradores realizaron biopsias renales en niños con antecedentes de SUH con proteinuria.(19) La mayoría de ellos presentaba esclerosis global y segmentaria con fibrosis intersticial. Los autores concluyeron que los niños con función renal normal y proteinuria leve presentan considerable daño renal y tienen riesgo de desarrollar insuficiencia renal tardíamente.(19)
Perlstein demostró que la carga oral de proteínas en 17 niños con antecedentes de SUH revelaba una reserva funcional renal disminuida en pacientes con antecedentes de SUH que tenían función renal y presión arterial normales, en comparación con niños sin tales antecedentes. Este estudio sugiere que la reserva funcional renal en niños con SUH puede estar disminuida a pesar de que la función renal y la presión se mantengan en valores normales.(26)
Los autores señalaron que no se conoce la importancia a largo plazo de este hallazgo y aún debe ser investigada, pero el estudio sugiere que la reserva funcional renal puede estar disminuida a pesar de la recuperación normal y que los niños con SUH requieren seguimiento prolongado. En el estudio de Spizzirri et al., 92,5% de los pacientes sin anuria se recuperaron, pero uno presentó proteinuria aislada, 2 tuvieron una disminución leve del índice de filtración glomerular (IFG) y uno sufrió insuficiencia renal terminal 19 años después de este episodio de SUH.(27) Además, 86% de los pacientes con proteinuria en el control de seguimiento del primer año tuvieron alteraciones renales al finalizar el seguimiento, pero un 25% de los pacientes sin proteinuria al año desarrollaron secuelas crónicas después de un lapso variable.
En resumen, los niños que aparentan haberse recuperado del SUH pueden presentar complicaciones tardías. Por lo tanto, es necesario determinar la fisiopatología de este síndrome para disminuir la mortalidad aguda y la morbilidad alargo plazo de esta enfermedad.
Mecanismos del daño ceñular mediado por la Shiga Toxina
Las toxinas shiga (también llamadas verotoxinas,verocitotoxinas [VT] o toxinas similares a la shiga [SLT]) están constituidas por dos subunidades: una subunidad A, de mayor tamaño, que inhibe la síntesis de proteínas mediante el bloqueo de la unión de aminocil -ARNt dependiente del factor-1 de elongación a los ribosomas y cinco subunidades B, más pequeñas, que intervienen en la unión de la toxina con un glucolípido de la membrana, la globotriaosilceramida o GB3.(28, 29) VT-1 producida por E. coli es casi idéntica a la shigatoxina producida típicamente por otros microorganismos gramnegativos, incluida Shigella, y sólo difiere por un aminoácido en la subunidad A; laVT-2 varía considerablemente de la shigatoxina y demuestra una homología de 55-60% aproximadamente.(28,29)
Las cepas de E. coli O157:H7aisladas de pacientes con SUH habitualmente producen VT-1 y VT-2 o sólo VT-2; las que producen sólo VT-1 son infrecuentes.(28,29) Es interesante señalar que una epidemia reciente de STEC 1 se acompañó de colitis hemorrágica en 526 niños y 35 adultos, pero no se asoció con SUH.(13) Aunque se detectaron anomalías leves en el análisis de orina de algunos pacientes, ninguno presentó síntomas típicos de SUH.(13) Este estudio epidemiológico resalta la importancia potencial de la VT-2 en el desarrollo de SUH en comparación con el papel de laVT-1.
Aunque no se demostraron niveles circulantes de VT libre en niños con SUH, se demostró que esta toxina atraviesa las células epiteliales gastrointestinales polarizadas a través de vías transcelulares.(30) Los estudios inmunohistoquímicos también demostraron la unión de las VT 1 y 2 a las células del epitelio tubular renal en un caso fatal de SUH típico con diarrea, lo que sugiere que la toxina se absorbe hacia la circulación sistémica, se une a las células renales y contribuye al daño celular.(31) La asociación entre STEC y SUH es muy firme, pero los mecanismos del daño celular mediado por esta toxina en el SUH aún son inciertos. Los estudios con VT-1 y células endoteliales de vena umbilical humana (HUVEC) demostraron que la VT tenía una acción tóxica directa en células endoteliales en crecimiento, no confluentes, pero la toxicidad era mucho menor en las células confluentes, en reposo.
Cuando las células HUVEC en reposo, resistentes a la VT, se trataron previamente con factor de necrosis tumoral-??(TNF-?) o interleuquina-1 (IL-1), se tornaron susceptibles a la toxicidad de la shigatoxina.(28,29) En otros estudios se demostró que la sensibilidad contra la VT de líneas celulares y tejidos específicos estaba relacionada con la expresión del receptor de GB3en la membrana y que las maniobras para aumentarla incrementan la sensibilidad a los efectos citotóxicos de la shigatoxina.(28,29) El receptor de la membrana plasmática GB3 regula la unión e internalización de la VT por endocitosis.
Estudios recientes en células endoteliales glomerulares humanas mostraron que éstas son sensibles a los efectos citotóxicos de la shigatoxina. En células de la microvasculatura glomerular altamente confluentes, la citotoxicidad mediada por VT requirió el tratamiento previo de la células con TNF?, que indujo aumento del número de receptores de GB3 en las células endoteliales glomerulares.(32) Cabe señalar que las VT 1 y 2 indujeron efectos citotóxicos similares sobre las células endoteliales de la microvasculatura glomerular. Por lo tanto, la fuerte asociación del SUH con las infecciones por STEC 2 no estaría relacionada con una mayor sensibilidad de las células endoteliales a la toxicidad mediada por VT-2 que a la VT-1.
Otros estudios muy interesantes demostraron que la VT puede influir considerablemente sobre la sensibilidad de algunas células a sus efectos citotóxicos sin un cambio significativo en la expresión del receptor GB3. En una serie de estudios, Lingwood y otros investigadores demostraron que, además de la concentración del receptor, la composición de ácidos grasos de GB3 y la longitud de la cadena fosfolipídica en la doble capa de fosfolípidos desempeña un papel importante en la distribución interna de la toxina y sus efectos citotóxicos ulteriores.(33-35) Cuando las células se sensibilizaron a la shiga toxina con butirato de sodio, el transporte intracelular retrógrado hacia la toxina internalizada unida al receptor se produjo hacia el retículo endoplásmico(RE) y la membrana nuclear más que hacia el aparato de Golgi, lo que se correlacionó con un marcado aumento de la sensibilidad celular a la shiga toxina.
La sensibilidad aumentada no se relacionó con mayor expresión de GB3 sino con receptores que contenían especies de ácidos grasos de cadena más corta y que dirigían la distribución intracelular de la toxina hacia la membrana nuclear. Esto explicaría la resistencia de algunas células a los efectos citotóxicos de la toxina a pesar de la expresión del receptor GB3 en la membrana plasmática.(35) Los investigadores sugirieron que las diferencias genéticas en la composición de ácidos grasos del receptorGB3 podrían desempeñar un papel en la susceptibilidad de los individuos al SUH cuando se exponen a la VT. En muchos de estos estudios, la inhibición de la síntesis proteica y la muerte celular fueron los determinantes de la lesión celular mediada por la VT.
Aunque algunas células responden a ésta con progresión a la muerte celular, otras no mueren y es probable que experimenten los efectos subletales de la VT. En células endoteliales aórticas bovinas, las VT 1 y 2 indujeron un aumento dependiente de la concentración de los niveles de transcripción del ARNm de preproendotelina-1 en concentraciones que tuvieron efectos menores sobre el ADN, el ARN y la síntesis proteica.(36) Por el contrario, la actividad de la enzima convertidora de Endotelina-1 y de la Oxido Nítrico Sintetasa constitutiva no fue alterada por las VT 1 o 2. Los autores sugirieron que las alteraciones en los mediadores vasculares derivados del endotelio mediadas por la VT podrían desempeñar un papel patogénico en el daño microvascular característico del SUH y la colitis hemorrágica.
También demostramos que las células endoteliales expuestas a la VT dañadas en niveles subletales son más susceptibles al daño mediado por los leucocitos polimorfonucleares activados que las células endoteliales no expuestas a la VT. (37) Es probable que la VT-1 y la VT-2 induzcan múltiples cambios subletales en las células que se recuperarán del daño .Aunque se considera que las células endoteliales son el blanco principal de la lesión mediada por la VT, estudios recientes demostraron que las células mesangiales, las células epiteliales tubulares renales, los monocitos y las líneas celulares derivadas de ellos son blanco de los efectos biológicos mediados por la ST y de su toxicidad.
Se demostró que las células mesangiales expresan los receptores GB3 y, al ser expuestas a la VT-1, exhiben inhibición de la síntesis proteica que se potencia con la preincubación con IL-1 o TNF?. Además, laVT1 indujo aumento de los niveles de ARNm deMCP-1 dependientes de la dosis y del tiempo(38) La incubación prolongada con shigatoxina-1produjo citotoxicidad sustancial en las células mesangiales. En otros estudios, las células mesangiales humanas expuestas a la VT-1 exhibieron disminución de la síntesis proteica, sin inducción de citoquinas o quimioquinas.(39) Las células del epitelio tubular renal exhiben diversas respuestas biológicas al exponerse a la VT.
Túbulos contorneados proximales humanos cultivados fueron sensibles a los efectos citotóxicos de la VT-1; este efecto fue mayor cuando las células se expusieron previamente a IL-1, lipopolisacárido y butirato, pero no lo fue cuando se preincubaron con TNF?.(40)Las concentraciones subletales de VT-1 indujeron aumento de la liberación de TNF??e IL-1 y mayor expresión de ARNm de TNF??e IL-1.(41)En otros estudios, la VT indujo apoptosis en una línea celular derivada de las células tubulares renales humanas.(42) Cabe señalar que se observó apoptosis de las células tubulares renales en especímenes patológicos de pacientes con SUH y se demostró que la VT se encontraba unida alas células tubulares distales en el SUH. (31,43)
Como se describe más adelante, también se demostró que las VT inducen múltiples respuestas biológicas de monocitos y macrófagos. Tomados en conjunto, estos estudios sugieren que las células mesangiales, las células epiteliales de los túbulos renales, los monocitos y los macrófagos son objetivos importantes del daño celular mediado por la VT en el SUH.
Las cepas de E. coli O157:H7aisladas de pacientes con SUH habitualmente producen VT-1 y VT-2 o sólo VT-2; las que producen sólo VT-1 son infrecuentes.(28,29) Es interesante señalar que una epidemia reciente de STEC 1 se acompañó de colitis hemorrágica en 526 niños y 35 adultos, pero no se asoció con SUH.(13) Aunque se detectaron anomalías leves en el análisis de orina de algunos pacientes, ninguno presentó síntomas típicos de SUH.(13) Este estudio epidemiológico resalta la importancia potencial de la VT-2 en el desarrollo de SUH en comparación con el papel de laVT-1.
Aunque no se demostraron niveles circulantes de VT libre en niños con SUH, se demostró que esta toxina atraviesa las células epiteliales gastrointestinales polarizadas a través de vías transcelulares.(30) Los estudios inmunohistoquímicos también demostraron la unión de las VT 1 y 2 a las células del epitelio tubular renal en un caso fatal de SUH típico con diarrea, lo que sugiere que la toxina se absorbe hacia la circulación sistémica, se une a las células renales y contribuye al daño celular.(31) La asociación entre STEC y SUH es muy firme, pero los mecanismos del daño celular mediado por esta toxina en el SUH aún son inciertos. Los estudios con VT-1 y células endoteliales de vena umbilical humana (HUVEC) demostraron que la VT tenía una acción tóxica directa en células endoteliales en crecimiento, no confluentes, pero la toxicidad era mucho menor en las células confluentes, en reposo.
Cuando las células HUVEC en reposo, resistentes a la VT, se trataron previamente con factor de necrosis tumoral-??(TNF-?) o interleuquina-1 (IL-1), se tornaron susceptibles a la toxicidad de la shigatoxina.(28,29) En otros estudios se demostró que la sensibilidad contra la VT de líneas celulares y tejidos específicos estaba relacionada con la expresión del receptor de GB3en la membrana y que las maniobras para aumentarla incrementan la sensibilidad a los efectos citotóxicos de la shigatoxina.(28,29) El receptor de la membrana plasmática GB3 regula la unión e internalización de la VT por endocitosis.
Estudios recientes en células endoteliales glomerulares humanas mostraron que éstas son sensibles a los efectos citotóxicos de la shigatoxina. En células de la microvasculatura glomerular altamente confluentes, la citotoxicidad mediada por VT requirió el tratamiento previo de la células con TNF?, que indujo aumento del número de receptores de GB3 en las células endoteliales glomerulares.(32) Cabe señalar que las VT 1 y 2 indujeron efectos citotóxicos similares sobre las células endoteliales de la microvasculatura glomerular. Por lo tanto, la fuerte asociación del SUH con las infecciones por STEC 2 no estaría relacionada con una mayor sensibilidad de las células endoteliales a la toxicidad mediada por VT-2 que a la VT-1.
Otros estudios muy interesantes demostraron que la VT puede influir considerablemente sobre la sensibilidad de algunas células a sus efectos citotóxicos sin un cambio significativo en la expresión del receptor GB3. En una serie de estudios, Lingwood y otros investigadores demostraron que, además de la concentración del receptor, la composición de ácidos grasos de GB3 y la longitud de la cadena fosfolipídica en la doble capa de fosfolípidos desempeña un papel importante en la distribución interna de la toxina y sus efectos citotóxicos ulteriores.(33-35) Cuando las células se sensibilizaron a la shiga toxina con butirato de sodio, el transporte intracelular retrógrado hacia la toxina internalizada unida al receptor se produjo hacia el retículo endoplásmico(RE) y la membrana nuclear más que hacia el aparato de Golgi, lo que se correlacionó con un marcado aumento de la sensibilidad celular a la shiga toxina.
La sensibilidad aumentada no se relacionó con mayor expresión de GB3 sino con receptores que contenían especies de ácidos grasos de cadena más corta y que dirigían la distribución intracelular de la toxina hacia la membrana nuclear. Esto explicaría la resistencia de algunas células a los efectos citotóxicos de la toxina a pesar de la expresión del receptor GB3 en la membrana plasmática.(35) Los investigadores sugirieron que las diferencias genéticas en la composición de ácidos grasos del receptorGB3 podrían desempeñar un papel en la susceptibilidad de los individuos al SUH cuando se exponen a la VT. En muchos de estos estudios, la inhibición de la síntesis proteica y la muerte celular fueron los determinantes de la lesión celular mediada por la VT.
Aunque algunas células responden a ésta con progresión a la muerte celular, otras no mueren y es probable que experimenten los efectos subletales de la VT. En células endoteliales aórticas bovinas, las VT 1 y 2 indujeron un aumento dependiente de la concentración de los niveles de transcripción del ARNm de preproendotelina-1 en concentraciones que tuvieron efectos menores sobre el ADN, el ARN y la síntesis proteica.(36) Por el contrario, la actividad de la enzima convertidora de Endotelina-1 y de la Oxido Nítrico Sintetasa constitutiva no fue alterada por las VT 1 o 2. Los autores sugirieron que las alteraciones en los mediadores vasculares derivados del endotelio mediadas por la VT podrían desempeñar un papel patogénico en el daño microvascular característico del SUH y la colitis hemorrágica.
También demostramos que las células endoteliales expuestas a la VT dañadas en niveles subletales son más susceptibles al daño mediado por los leucocitos polimorfonucleares activados que las células endoteliales no expuestas a la VT. (37) Es probable que la VT-1 y la VT-2 induzcan múltiples cambios subletales en las células que se recuperarán del daño .Aunque se considera que las células endoteliales son el blanco principal de la lesión mediada por la VT, estudios recientes demostraron que las células mesangiales, las células epiteliales tubulares renales, los monocitos y las líneas celulares derivadas de ellos son blanco de los efectos biológicos mediados por la ST y de su toxicidad.
Se demostró que las células mesangiales expresan los receptores GB3 y, al ser expuestas a la VT-1, exhiben inhibición de la síntesis proteica que se potencia con la preincubación con IL-1 o TNF?. Además, laVT1 indujo aumento de los niveles de ARNm deMCP-1 dependientes de la dosis y del tiempo(38) La incubación prolongada con shigatoxina-1produjo citotoxicidad sustancial en las células mesangiales. En otros estudios, las células mesangiales humanas expuestas a la VT-1 exhibieron disminución de la síntesis proteica, sin inducción de citoquinas o quimioquinas.(39) Las células del epitelio tubular renal exhiben diversas respuestas biológicas al exponerse a la VT.
Túbulos contorneados proximales humanos cultivados fueron sensibles a los efectos citotóxicos de la VT-1; este efecto fue mayor cuando las células se expusieron previamente a IL-1, lipopolisacárido y butirato, pero no lo fue cuando se preincubaron con TNF?.(40)Las concentraciones subletales de VT-1 indujeron aumento de la liberación de TNF??e IL-1 y mayor expresión de ARNm de TNF??e IL-1.(41)En otros estudios, la VT indujo apoptosis en una línea celular derivada de las células tubulares renales humanas.(42) Cabe señalar que se observó apoptosis de las células tubulares renales en especímenes patológicos de pacientes con SUH y se demostró que la VT se encontraba unida alas células tubulares distales en el SUH. (31,43)
Como se describe más adelante, también se demostró que las VT inducen múltiples respuestas biológicas de monocitos y macrófagos. Tomados en conjunto, estos estudios sugieren que las células mesangiales, las células epiteliales de los túbulos renales, los monocitos y los macrófagos son objetivos importantes del daño celular mediado por la VT en el SUH.
Papel de la respuesta inflamatoria en el SUH
Las VT tienen efectos directos sobre las células endoteliales, mesangiales y epiteliales, pero es muy probable que los mediadores inflamatorios también sean importantes en la patogenia del daño endotelial.(44-46) Los estudios en animales demostraron que los macrófagos son mediadores importantes en el daño celular inducido por la VT y que ésta induce la expresión de TNF en ratones; otras investigaciones en animales comprobaron que también aumenta la producción de óxido nítrico en macrófagos de ratones expuestos a la VT. (47,48)
Los monocitos humanos y las líneas celulares derivadas de ellos responden a la VT con aumento de la secreción de citoquinas inflamatorias, incluidos TNFa, IL-1 e IL-6.(18,49) El aumento de la expresión de ARNm de TNFa fue precedido por la translocación nuclear de los activadores de la transcripción NF-kB y AP-1.(50) Se describieron niveles elevados de IL-8 yTNF, así como signos de estrés oxidativo en niños con SUH, lo que sugiere que la respuesta inflamatoria y, en particular de PMN, son mediadores muy importantes en el daño microvascular del SUH. (44-46)
Además, recientemente se observó que los niveles urinarios de MCP-1 eIL-8 estaban significativamente elevados en niños durante la fase aguda del SUH. (18) Varios estudios sugieren un papel de la lesión oxidante en la patogenia del SUH. En primer lugar, un recuento elevado de PNM es típico de la enfermedad; un recuento elevado de leucocitos se asocia con enfermedad más grave y de peor evolución.(45) En segundo lugar, los niveles sanguíneos de interleuquina-8, una citoquina que estimula la desgranulación de los PMN y la generación de oxidantes, están elevados en niños con SUH.
La enzima elastasa, que provoca la desgranulación de los PMN formando complejos con la alfa-1-antitripsina, está elevada en niños durante la fase aguda del SUH y en modelos animales de la enfermedad.(45,46) En estudios in vitro, los PMN fueron más adherentes a las HUVEC expuestas a laVT-1.(46) Demostramos que las células endoteliales de vena umbilical humana y las células endoteliales glomerulares humanas expuestas a TNF y VT son más sensibles al daño celular mediado por PMN que las células endoteliales que no habían sido expuestas a la toxina.(37)
Estos estudios sugieren que las células endoteliales expuestas a la toxina son más sensibles al daño mediado por los PMN y que la exposición a la VT provoca amplificación del daño endotelial mediado por los PMN. Tomados en conjunto, estos estudios sugieren un papel importante de los oxidantes y las proteasas derivadas de los PMN activados en la patogenia del SUH.
En consecuencia, la VT provoca daño letal y subletal directo en las células del endotelio glomerular y estimula la producción de citoquinas para inducir la activación de PMN, lo que conduce a la amplificación del daño endotelial por mecanismos mediados por factores oxidantes o por proteasas . La célula endotelial dañada altera sus características tromborresistentes normales y se vuelve trombogénica, con inicio del depósito de trombos de plaquetas en la microvasculatura.
Los monocitos humanos y las líneas celulares derivadas de ellos responden a la VT con aumento de la secreción de citoquinas inflamatorias, incluidos TNFa, IL-1 e IL-6.(18,49) El aumento de la expresión de ARNm de TNFa fue precedido por la translocación nuclear de los activadores de la transcripción NF-kB y AP-1.(50) Se describieron niveles elevados de IL-8 yTNF, así como signos de estrés oxidativo en niños con SUH, lo que sugiere que la respuesta inflamatoria y, en particular de PMN, son mediadores muy importantes en el daño microvascular del SUH. (44-46)
Además, recientemente se observó que los niveles urinarios de MCP-1 eIL-8 estaban significativamente elevados en niños durante la fase aguda del SUH. (18) Varios estudios sugieren un papel de la lesión oxidante en la patogenia del SUH. En primer lugar, un recuento elevado de PNM es típico de la enfermedad; un recuento elevado de leucocitos se asocia con enfermedad más grave y de peor evolución.(45) En segundo lugar, los niveles sanguíneos de interleuquina-8, una citoquina que estimula la desgranulación de los PMN y la generación de oxidantes, están elevados en niños con SUH.
La enzima elastasa, que provoca la desgranulación de los PMN formando complejos con la alfa-1-antitripsina, está elevada en niños durante la fase aguda del SUH y en modelos animales de la enfermedad.(45,46) En estudios in vitro, los PMN fueron más adherentes a las HUVEC expuestas a laVT-1.(46) Demostramos que las células endoteliales de vena umbilical humana y las células endoteliales glomerulares humanas expuestas a TNF y VT son más sensibles al daño celular mediado por PMN que las células endoteliales que no habían sido expuestas a la toxina.(37)
Estos estudios sugieren que las células endoteliales expuestas a la toxina son más sensibles al daño mediado por los PMN y que la exposición a la VT provoca amplificación del daño endotelial mediado por los PMN. Tomados en conjunto, estos estudios sugieren un papel importante de los oxidantes y las proteasas derivadas de los PMN activados en la patogenia del SUH.
En consecuencia, la VT provoca daño letal y subletal directo en las células del endotelio glomerular y estimula la producción de citoquinas para inducir la activación de PMN, lo que conduce a la amplificación del daño endotelial por mecanismos mediados por factores oxidantes o por proteasas . La célula endotelial dañada altera sus características tromborresistentes normales y se vuelve trombogénica, con inicio del depósito de trombos de plaquetas en la microvasculatura.
Repercusiones terapéuticas y conclusión
El mayor conocimiento de los mecanismos del daño celular mediado por la VT en el SUH tiene importantes repercusiones en el manejo actual de la enfermedad y en los tratamientos futuros. Algunos estudios demostraron un efecto perjudicial del tratamiento con antibióticos en la colitis hemorrágica. En niños con colitis hemorrágica asociada con STEC tratados con antibióticos se observó que desarrollaban SUH con mayor frecuencia que los que no recibieron este tratamiento.(51)
Otros estudios no confirmaron tal asociación y un metaanálisis reciente concluyó que la administración de antibióticos a personas infectadas con STEC no se asociaba con el desarrollo de SUH. (52,53) Aunque no hay consenso sobre el efecto beneficioso o perjudicial de los antibióticos en la infección por STEC, estudios in vitro demostraron que las concentraciones subinhibitorias de varios agentes antimicrobianos promueven y aumentan la liberación de VT. (54)
Se desarrolló un compuesto de sílice diamídico de diatomea unido a una cadena de oligosacárido (Synsorb Pk) que se une ávidamente a la shigatoxina y la neutraliza. Recientemente se completó un ensayo clínico para determinar si la administración oral de Synsorb Pk puede disminuirla velocidad de progresión de la colitis hemorrágica a SUH, la necesidad de diálisis o de complicaciones extrarrenales en niños con SUH.(55,56) Lamentablemente, el Synsorb Pk no resultó útil para prevenir las complicaciones extrarrenales o disminuir la duración de la diálisis en niños con SUH de comienzo reciente. E
l Starfish es un nuevo compuesto desarrollado recientemente que se une a la shiga toxina con una eficiencia 1.000 veces mayor que Synsorb Pk. Starfish es un oligosacárido pentavalente que fija shigatoxina y puede ser administrado por vía intravenosa.(57) Recientemente(58) se está desarrollando otro producto llamado Daisy, con la misma base fisiopatogénica, que utilizado en ratones da resultados muy promisorios. Es posible que la mejor comprensión de la fisiopatología del daño celular en el SUH permita desarrollar nuevas estrategias terapéuticas para los niños afectados y prevenir la mortalidad aguda y la morbilidad a largo plazo de la enfermedad.
Conclusiones:
El SUH típico con diarrea se asocia firmemente con la colitis hemorrágica por STEC, lo que sugiere que estas toxinas son muy importantes en la fisiopatología del SUH. Las VT se unen al receptor GB3 en la membrana plasmática e inician su acción citotóxica. Estudios recientes demostraron que las alteraciones en el contenido lipídico del receptor GB3 y en la distribución intracelular de la toxina son determinantes importantes de la susceptibilidad de las células a la citotoxicidad mediada por VT.
Otros estudios demostraron que las VT inician alteraciones biológicas en células dañadas subletalmente y que las células mesangiales, del epitelio tubular renal, monocitos y las líneas celulares derivadas de ellos son blancos de la lesión celular mediada por VT. También se informó que la respuesta inflamatoria y la activación de los PMN desempeñarían un papel importante en el daño celular mediado por VT. Estos estudios tienen importantes consecuencias para los tratamientos actuales y futuros del SUH.
Agradecimientos: Esta revisión y el trabajo en el laboratorio de los autores fueron posibles gracias a la subvención DK52175 del NIH.
Otros estudios no confirmaron tal asociación y un metaanálisis reciente concluyó que la administración de antibióticos a personas infectadas con STEC no se asociaba con el desarrollo de SUH. (52,53) Aunque no hay consenso sobre el efecto beneficioso o perjudicial de los antibióticos en la infección por STEC, estudios in vitro demostraron que las concentraciones subinhibitorias de varios agentes antimicrobianos promueven y aumentan la liberación de VT. (54)
Se desarrolló un compuesto de sílice diamídico de diatomea unido a una cadena de oligosacárido (Synsorb Pk) que se une ávidamente a la shigatoxina y la neutraliza. Recientemente se completó un ensayo clínico para determinar si la administración oral de Synsorb Pk puede disminuirla velocidad de progresión de la colitis hemorrágica a SUH, la necesidad de diálisis o de complicaciones extrarrenales en niños con SUH.(55,56) Lamentablemente, el Synsorb Pk no resultó útil para prevenir las complicaciones extrarrenales o disminuir la duración de la diálisis en niños con SUH de comienzo reciente. E
l Starfish es un nuevo compuesto desarrollado recientemente que se une a la shiga toxina con una eficiencia 1.000 veces mayor que Synsorb Pk. Starfish es un oligosacárido pentavalente que fija shigatoxina y puede ser administrado por vía intravenosa.(57) Recientemente(58) se está desarrollando otro producto llamado Daisy, con la misma base fisiopatogénica, que utilizado en ratones da resultados muy promisorios. Es posible que la mejor comprensión de la fisiopatología del daño celular en el SUH permita desarrollar nuevas estrategias terapéuticas para los niños afectados y prevenir la mortalidad aguda y la morbilidad a largo plazo de la enfermedad.
Conclusiones:
El SUH típico con diarrea se asocia firmemente con la colitis hemorrágica por STEC, lo que sugiere que estas toxinas son muy importantes en la fisiopatología del SUH. Las VT se unen al receptor GB3 en la membrana plasmática e inician su acción citotóxica. Estudios recientes demostraron que las alteraciones en el contenido lipídico del receptor GB3 y en la distribución intracelular de la toxina son determinantes importantes de la susceptibilidad de las células a la citotoxicidad mediada por VT.
Otros estudios demostraron que las VT inician alteraciones biológicas en células dañadas subletalmente y que las células mesangiales, del epitelio tubular renal, monocitos y las líneas celulares derivadas de ellos son blancos de la lesión celular mediada por VT. También se informó que la respuesta inflamatoria y la activación de los PMN desempeñarían un papel importante en el daño celular mediado por VT. Estos estudios tienen importantes consecuencias para los tratamientos actuales y futuros del SUH.
Agradecimientos: Esta revisión y el trabajo en el laboratorio de los autores fueron posibles gracias a la subvención DK52175 del NIH.
Anemia falciforme
Anemia falciforme
Actualización sobre anemia de células falciformes en pacientes pediátricosAutor: Frédéric B. Piel, Martin H. Steinberg, and David C. Rees N Engl J Med 2017;376:1561-73.
Introducción
La enfermedad de células falciformes es un problema de salud mundial cada vez mayor. Las estimaciones sugieren que cada año aproximadamente 300.000 niños nacen con anemia de células falciformes, que se define por la homocigosidad para el gen de la hemoglobina falciforme (HbS) (es decir, para una mutación con cambio de sentido o missense [Glu6Val, rs334] en el gen β-globina [HBB]) y que este número podría llegar a 400.000 en el 2050.
Aunque el diagnóstico precoz, la profilaxis con penicilina, las transfusiones de sangre, la imagen Doppler transcraneal, la hidroxiurea y el trasplante de células madre hematopoyéticas pueden mejorar dramáticamente la sobrevida y la calidad de vida de los pacientes con enfermedad de células falciformes, la comprensión del rol de los factores genéticos y no genéticos en la explicación de la diversidad fenotípica de esta enfermedad mendeliana sigue siendo limitada.
La mejor predicción de la gravedad de la anemia falciforme podría conducir a un tratamiento y manejo más precisos.Más allá de los modificadores bien conocidos de la gravedad de la enfermedad, como los niveles de hemoglobina fetal (HbF) y la α-talasemia, otras variantes genéticas pueden afectar a sub-fenotipos específicos.
Del mismo modo, aunque la influencia de la altitud y la temperatura se ha visto reflejadalargamente en el asesoramiento a pacientes con enfermedad de células falciformes, estudios recientes de factores no genéticos, incluyendo el clima y la calidad del aire, sugieren asociaciones más complejas entre los factores ambientales y las complicaciones clínicas. Nuevos tratamientos y estrategias de manejo abocados a estos factores genéticos y no genéticos podrían mejorar sustancial y rápidamente la calidad de vida y reducir los costos del cuidado de la salud de los pacientes con enfermedad de células falciformes.
► Distribución y carga de enfermedad
La enfermedad de células falciformes es el trastorno monogénico más frecuente. La prevalencia de la enfermedad es alta en grandes áreas del África subsahariana, la cuenca del Mediterráneo, el Oriente Medio y la India, debido al notable nivel de protección que el rasgo de células falciformes (es decir, la heterocigosidad para la mutación de células falciformes en HBB) proporciona contra la malaria severa.
Aunque el papel exacto de los diversos mecanismos de protección que se han identificado todavía se está debatiendo, la "hipótesis de la malaria" formulada por Haldane en 1949 y por Allison en 1954 es un ejemplo de selección natural y polimorfismo equilibrado, un proceso en curso. Debido al comercio de esclavos y a los movimientos de población contemporáneos, la distribución de la enfermedad de células falciformes se ha extendido mucho más allá de sus orígenes.
Las estimaciones de población en los Estados Unidos sugieren que un total de aproximadamente 100.000 personas tienen la enfermedad. No existe una estimación anual confiable para ningún otro país ni una estimación global, pero las estimaciones para recién nacidos sugieren consistentemente que nacen 300.000 bebés por año con anemia de células falciformes. La gran mayoría de estos nacimientos ocurren en tres países: Nigeria, la República Democrática del Congo, y la India.
El número de pacientes con enfermedad de células falciformes se espera que aumente, tanto en los países de altos ingresos como en los países de ingresos más bajos. En los países de altos ingresos, este aumento refleja en gran medida las ganancias en la expectativa de vida de las personas afectadas como resultado de intervenciones tales como la detección en recién nacidos, la profilaxis con penicilina, la prevención primaria del accidente cerebrovascular, y el tratamiento con hidroxiurea. La esperanza de vida ha mejorado significativamente en los países de altos ingresos en los últimos 40 años, con una mortalidad infantil ahora cercana a la de la población general y una sobrevida mediana observada de más de 60 años.
A pesar de estos notables logros, la expectativa de vida de los pacientes con anemia de células falciformes se reduce en unos 30 años, incluso con los mejores cuidados médicos, y la calidad de vida es pobre. El tratamiento con hidroxiurea - la única terapia farmacológica aprobada para la enfermedad de células falciformes - se utiliza cada vez más en adultos y niños.
Sin embargo, el tratamiento y el manejo de la enfermedad siguen siendo costosos, haciendo que el pleno acceso a la atención esté disponible sólo para los más privilegiados; de otra manera, el acceso es muy limitado debido a las crecientes presiones sobre los servicios de salud pública. Se destacan nuevos desarrollos en el manejo de la anemia falciforme a través de recientes y continuosensayos clínicos de fase 3, y por el creciente número de pacientes que se están beneficiando con el trasplante de células madre hematopoyéticas.
En los países de menores ingresos, donde la mortalidad infantil por todas las causas se ha reducido en las últimas dos décadas, un mayor número de lactantes y niños pequeños afectados sobreviven hasta la edad adulta, requiriendo diagnóstico y tratamiento.
En África, donde hay una falta de detección neonatal y de vacunación infantil rutinaria y donde la malaria, la desnutrición y la pobreza siguen siendo importantes retos, la mortalidad entre los niños con enfermedad de células falciformes menores de 5 años de edad puede ser tan alta como del 90%. Aunque algunos programas de detección a gran escala han sido lanzados con relativo éxito recientemente, la falta de una infraestructura sanitaria básica en muchas regiones hace que la prevención y el manejo de la enfermedad de células falciformes sea difícil.
► Fisiopatología
El síndrome torácico agudo es un ejemplo típico de fracaso orgánico en la enfermedad de células falciformes y una de las principales causas de hospitalización y muerte entre los pacientes
La enfermedad de células falciformes es un trastorno multisistémico causado por una sola mutación genética. Casi todos lo sórganos del cuerpo pueden verse afectados. Caracterizada por la presencia de eritrocitos anormales dañados por la HbS, esta variante de la hemoglobina adulta normal (HbA) se hereda bien de ambos padres (homocigosidad para el gen HbS) o de un solo padre, junto con otra variante de la hemoglobina, como la hemoglobina C (HbC), o con Β-talasemia (heterocigosidad compuesta).
Cuando está desoxigenada, la HbS se polimeriza, dañando al eritrocito y haciendo que pierdacationes yagua. Estas células dañadas tienen anomalías en sus características reológicas y en la expresión de moléculas de adhesión, resultando en anemia hemolíticay en la probabilidad de bloqueo de los pequeños vasos sanguíneos, causando a su vez vaso-oclusión. La vaso-oclusión típicamente causa complicaciones agudas, incluyendo daño isquémico a los tejidos, dando como resultado dolor severo o falla orgánica. El síndrome torácico agudo es un ejemplo típico de fracaso orgánico en la enfermedad de células falciformes y una de las principales causas de hospitalización y muerte entre los pacientes.
Aunque la polimerización de la HbS, la vaso-oclusión, y la anemia hemolítica son centrales en la fisiopatología de la enfermedad de células falciformes, precipitan una cascada de eventos patológicos que, a su vez, desencadenan una amplia gama de complicaciones. Estos procesos incluyen:
- disfunción vascular-endotelial
- deficiencia funcional del óxido nítrico
- inflamación
- estrés oxidativo
- lesión por reperfusión
- hipercoagulabilidad
- aumento de la adhesividad de los neutrófilos
- activación plaquetaria
La interacción y la importancia relativa de estos trastornos son pobremente entendidos y probablemente difieren según la complicación particular. Las complicaciones crónicas se dividen en dos grupos principales:
- Las relacionadas con vasculopatía de grandes vasos (enfermedad cerebrovascular, hipertensión pulmonar, priapismo y retinopatía).
- Las causadas por daño orgánico isquémico progresivo(hipoesplenismo, insuficiencia renal, enfermedad ósea, y daño hepático). El hipoesplenismo es una causa particularmente importante de enfermedad y muerte enniños pequeños debido al mayor riesgo deinfección.
Los pacientes con anemia falciforme pueden tener cualquiera de una serie de genotipos de hemoglobina. Casi todos los estudios genéticos de la enfermedad falciforme se han concentrado sobre el genotipo de la anemia drepanocítica (es decir, HBB Glu6Val, rs334).
Otros genotipos de la enfermedad de células falciformes son debidos a heterocigocidad compuesta para el gen HbS y otras variantes de la hemoglobina (Hb) tales como HbC, HbE y HbD o a diversas variedades de HbS-β-talasemia.
Con excepción de la HbS-β0-talasemia (β0 denota sin HbA), los genotipos heterocigotas compuestos de la enfermedad de células falciformes suelen ser menos clínicamente severos que el genotipo de la anemia drepanocítica. Sin embargo, dentro de cada genotipo de enfermedad existe una heterogeneidad fenotípica sustancial.
Muchos estudios han investigado las relaciones fenotipo-genotipo en la enfermedad de células falciformes. La evidencia previa de estudios de pacientes con enfermedad de células falciformes expuestos a alta altitud destacó la influencia de los factores medio ambientales en las complicaciones de la enfermedad.
Posteriormente, la identificación genética de varios haplotipos del gen HbS (Bantu, Benin, Camerún, Senegal y Arábigo-Indio), sugiriendo diferentes orígenes de la mutación HbS entre las áreas de alta prevalencia, llevó a la especulación de que el haplotipo del gen HbS podría explicar las diferencias fenotípicas.
Un estudio piloto, observando a nueve pares de gemelos idénticos, trató de desentrañar los roles de los factores genéticos y no genéticos, con resultados interesantes pero limitados debido al pequeño tamaño de la muestra. Las siguientes secciones resumen el conocimiento actual del rol de los modificadores genéticos y no genéticos.
► Modificadores genéticos de la severidad de la enfermedad
La diversidad fenotípica de la anemia falciforme es parcialmente explicada por variantes genéticas que controlan la expresión de los genes de HbF y por la co-inheritancia del gen de la α-talasemia. El rol de otros potenciales modificadores genéticos es menos claro.
♦ α-talasemia
La polimerización de la HbS desoxigenada inicia los cambios patológicos que caracterizan a la enfermedad de células falciformes. La velocidad de polimerización de la HbS es altamente dependiente del nivel de hemoglobina de los eritrocitos, con niveles más bajos de HbS conduciendo a menor daño celular; la α-talasemia reduce el nivel de hemoglobinaen la célula, mitigando indirectamente el daño eritrocitario inducido por el polímero de HbS. Causada más a menudo por la supresión de uno o dos de los cuatro genes de α-globina, la α-talasemia está presente en un tercio de los pacientes de origen africano y en casi la mitad de los pacientes de ascendencia de Oriente Medio o India.
La co-inheritancia de α-talasemia y anemia drepanocítica se caracteriza por mayores niveles de hemoglobina que en la herencia de la anemia falciforme sola, así como por un menor volumen corpuscular medio, menos hemólisis y menos complicaciones que han sido asociadas epidemiológicamente con la hemólisis.
A la inversa, algunas características de la enfermedad asociadas con la vaso-oclusión celular falciforme, como los episodios dolorosos agudos, son más comunes en la anemia falciforme y la α-talasemia co-heredadas, tal vez debido al mayor volumen de células aglomeradas.
La homeostasis vascular es mantenida por el óxido nítrico endotelial, que relaja el músculo liso perivascular. Se planteó la hipótesis de que la reducción de algunas complicaciones asociadas con la hemólisis tanto en pacientes con anemia de células falciformes como con α-talasemia en parte era resultado de la preservación de la bio-disponibilidad del óxido nítrico que se ve comprometido por la hemólisis intravascular de los drepanocitos.
Durante la hemólisis, la hemoglobina liberada en el plasma reacciona con el óxido nítrico formando nitrato inerte, y la arginasa eritrocitaria se metaboliza a arginina, el sustrato para las óxido nítrico sintetasas. La actividad del óxido nítrico también se inhibe por reacción con la dimetilarginina asimétrica. La bio-disponibilidad del óxido nítrico contribuye a la variabilidad fenotípica de la anemia falciforme más allá de la co-inheritancia de las α-talasemias.
♦ Hemoglobina fetal
La HbF interrumpe la polimerización de la HbS, ya que la HbF es excluida del polímero de HbS. Los niveles de HbF alcanzan su punto máximo a mitad de la gestación; al momento que un niño sano y no afectado llega a la edad de 6 meses, la HbF representa menos del 1% de la hemoglobina total, pero los niveles son mayores en la mayoría de los adultos con enfermedad de células falciformes.
La primera variante genética asociada con un aumento de la HbF en la anemia de células falciformes, marcador del haplotipo de Senegal del grupo HBB, fue un polimorfismo de un solo nucleótido (PSN) (rs7482144) en la región promotora del HBG2, uno de los genes HbF pareados. Los portadores de este haplotipo tenían niveles de HbF de aproximadamente 10%, en comparación con el 5 a 6% en los portadores de los otros dos haplotipos africanos comunes.
El silenciamiento de los genes de HbF desde el desarrollo fetal hasta la adultez se explica por la actividad del BCL11A y del ZBTB7A. La variación genética de un potenciador eritrocitario específico del BCL11A, junto con polimorfismos en un potenciador del MYB, explican el 10 al 50% de la varianza de la HbF observada entre las personas con anemia drepanocítica, dependiendo de la población examinada.
En la Provincia Oriental de Arabia Saudita y en la India, el gen de la HbS es a menudo un haplotipo HBB árabe-indio de origen autóctono. En estos casos, los niveles de HbF en adultos son casi dos veces los encontrados en el haplotipo senegalés. En consecuencia, la enfermedad, especialmente en la infancia, cuando los niveles de HbF son de alrededor del 30%, suele ser más moderada.
La base genética de los altos niveles de HbF en estas personas podría en parte derivar en polimorfismos haplotipo-específicos del super potenciador del grupo HBB y otras variantes exclusivas de este haplotipo. Los pacientes de Arabia Saudita con el haplotipo de Benin tienen niveles de HbF que son casi el doble de los niveles en pacientes africanos con el mismo haplotipo. La razón de esta diferencia es desconocida.
La HbF no mejora todos los subfenotipos de la enfermedad en la misma medida. El determinante crítico del efecto de la HbF sobre el fenotipo de la enfermedad de células falciformes es su nivel en cada eritrocito.En los heterocigotos compuestos para HbS y persistencia hereditaria de HbF, donde el HBB está suprimido, la HbF supone aproximadamente el 30% de la hemoglobina total y se distribuye homogéneamente en la población de glóbulos rojos, con cada célula conteniendo alrededor de 10 pg.
Este nivel es suficiente para evitar la polimerización de la HbS desoxigenada, de modo que las personas con este genotipo tienen niveles de hemoglobina casi normales y son en su mayoría asintomáticos. Aunque la hidroxiurea aumenta los niveles de HbF en la mayoría de los pacientes, su distribución en eritrocitos falciformes es heterogénea.
Las células con niveles inferiores de HbF están menos protegidas de los daños inducidos por el polímero, persiste la anemia hemolítica y la mayoría de los pacientes permanecen sintomáticos, aunque con una tasa reducida de complicaciones y tal vez una sobrevida mejorada. Junto con la hidroxiurea, varios nuevos tratamientos basados en la inducción de HbF (por ejemplo, inhibidores de la histona desacetilasa, inhibidores de la histona específica de lisina desmetilasa 1 [LSD1], e inmunomoduladores) se encuentran actualmente en diversas fases de investigación.
► Otros modificadores genéticos
La complejidad biológica de la anemia falciforme proporciona numerosos sitios para su modulación genética por genes cuyas acciones primarias son extraeritrocíticas. Muchos polimorfismos genéticos han sido asociados con sub-fenotipos específicos, con un efecto protector o permisivo sobre la característica biológica de interés.Se han encontrado marcadores de asociación más claros para el accidente cerebrovascular (ACV).
Se genotiparon treinta y ocho PSNs en 22 genes de 130 pacientes con anemia de células falciformes y ACV y de 103 pacientes que presentaban anemia falciforme sin complicaciones (controles). Además de la conocida asociación de la α-talasemia con un riesgo reducido de ACV, los PSNs en ANXA2, TEK, ADCY9 yTGFBR3 se asociaron con un aumento o una disminución del riesgo de ACV. Estos resultados confirmaron parcialmente los resultados de un estudio que evaluó 108PSNs en 39 genes candidatos y que mostró que 31PSNs en 12 genes modularon el riesgo de ACV. Otros marcadores genéticos incluyen polimorfismos en los genes para los subfenotipos de bacteriemia, osteonecrosis, y priapismo (CCL5, BMP6 y KL, respectivamente).
Un resultado consistente de los estudios que preseleccionaron genes candidatos para probar la asociación de sus variantes con subfenotipos múltiples fue la detección de asociaciones con varios genes de la vía de proteínas morfogenéticas óseas SMAD, que incluye al factor de crecimiento transformante β. Esta vía regula diversos procesos celulares que son importantes en la fisiopatología de la anemia falciforme, incluyendo inflamación, fibrosis, proliferación celular y hematopoyesis, osteogénesis, angiogénesis, cicatrización de heridas y respuesta inmune.Los estudios de asociación a nivel genómico, que proporcionan una evaluación imparcial de la asociación genética con un fenotipo, no han replicado estos resultados basados en genes candidatos.
Tales estudios requieren miles de participantes y un cuidadoso análisis fenotípico para alcanzar significación estadística si la contribución de una variante genética a un fenotipo es pequeña. Para la enfermedad por anemia falciforme, la obtención de una muestra tan grande no ha sido posible.Aparte de los resultados de los estudios que usan el nivel de HbF como un subfenotipo, los estudios de asociación genómica han contribuido hasta ahora poco a la comprensión de las bases genéticas de la heterogeneidad fenotípica en la enfermedad de células falciformes.
► Modificadores no genéticos de la severidad de la enfermedad
La investigación se ha centrado principalmente en las variantes genéticas que interfieren en la variabilidad fenotípica de la anemia de células falciformes, y el rol de los factores no genéticos ha sido relativamente descuidado. Sin embargo, los factores no genéticos pueden explicar gran parte de la variabilidad clínica.
Más dramáticamente, la sobrevida de los niños con enfermedad de células falciformes en los países de altos ingresos se aproxima a la de los niños no afectados, mientras que en la mayor parte del África subsahariana hasta el 90% de los niños con esta enfermedad mueren, incluso pese a que estas poblaciones son genéticamente muysimilares. Los factores no genéticos incluyen el clima y la calidad del aire, así como los factores socioeconómicos, que se evalúan, por ejemplo, en base al acceso a la atención médica, las transfusiones de sangre seguras, y el tratamiento de las infecciones.
► Factores climáticos y meteorológicos
Una relación entre el clima frío y las complicaciones agudas de la enfermedad de células falciformes se describió por primera vez en Estados Unidos en 1924. Los mecanismos propuestos incluyen un clima frío que causa aumento de las infecciones y vasoconstricción periférica que lleva a mayor desoxigenación con disminución de la presión de flujo y efectos por secuestro vascular. Sin embargo, el vínculo entre el clima frío y el dolor agudo se identificó de forma inconsistente en análisis de series por tiempos más prolongados.
Estudios realizados en Ghana, Nueva York, Virginia, Jamaica, Kuwait, y Canadá sugieren un vínculo entre el frío y el dolor a lo largo de un rango declimas. Por el contrario, no se hallaron efectos del clima frío en Chicago y Atlanta y en dos estudios separados llevados a cabo en Londres. Un reciente estudio en París mostró que tanto el clima cálido como frío se asociaron con aumento de los episodios de dolor.
Estos hallazgos inconsistentes pueden reflejar diferencias en los métodos y análisis utilizados en estos estudios, que se limitaron todos a analizar el número de ingresos hospitalarios, que es un sustituto muy indirecto de los cambios fisiopatológicos asociados con las variaciones de temperatura. Algunas de las inconsistencias pueden también reflejar la influencia de las características específicas de la ubicación, incluyendo vivienda, vestimenta y factores sociales y geográficos, sobre los efectos de la temperatura.
Aunque no suele ser notado por los pacientes, la velocidad del viento ha surgido como un factor consistentemente asociado con dolor en la anemia de células falciformes, y la mayor velocidad del viento ha sido relacionada con una mayor tasa de hospitalizaciones por dolor en Inglaterra, Francia, Canadá, y Estados Unidos. No está claro cómo las altas velocidades del viento podrían precipitar episodios de dolor agudo, aunque existen pruebas de que el enfriamiento de la piel podría provocar vaso-oclusión posiblemente como resultado de una alteración del control del tono vascular.
Tanto la humedad alta como la baja han sido asociadas con un aumento de los ingresos hospitalarios por dolor, y se observaron mayores puntuaciones de dolor conel aumento de la humedad en un estudio en Canadá. Se reportan aumentos de los episodios de dolor agudo durante la estación lluviosa en regiones con climas tropicales, tales como Jamaica y Nigeria, aunque no emergen efectos consistentes por la lluvia donde el clima es templado, como Francia e Inglaterra. Nuevamente, las inconsistencias pueden deberse a diferencias en el hábitat y en los factores sociales.
► Calidad del aire
La contaminación atmosférica está emergiendo como una causa importante de enfermedad, aunque su papel en la enfermedad de células falciformes es poco comprendido. En Europa y en los Estados Unidos, los pacientes con anemia falciforme viven predominantemente en zonas urbanas, donde están expuestos a concentraciones elevadas de contaminantes, incluyendo varios con bioactividad. Como se discutió anteriormente, la enfermedad de células falciformes se asocia con deficiencia funcional de óxido nítrico.
Pequeños estudios retrospectivos en Londres sugirieron que la exposición a corto plazo a mayores niveles de óxido nítrico atmosférico se asoció con menos ingresos hospitalarios y que la exposición prolongada se asoció con disminución de los marcadores de hemólisis.
El monóxido de carbono es otro componente bioactivo, contaminante gaseoso, que en teoría puede tener un beneficio terapéutico en la enfermedad de células falciformes, ya que la carboxihemoglobina está bloqueada en la forma R (relajada) y no puede polimerizar. El rol terapéutico del monóxido de carbono está siendo explorado actualmente en un ensayo de carboxihemoglobina bovina pegilada. Estudios en París y Londres demostraron que mayores niveles atmosféricos de monóxido de carbono se asociaron con disminución de los ingresos hospitalarios por dolor agudo, aunque se observó el efecto contrario en São Paulo.
Otros contaminantes potencialmente importantes incluyen al ozono (O3), óxidos de nitrógeno (NO y NO2), óxidos de azufre (SO y SO2), y material particulado (PM10 y PM2,5 [es decir, material particulado con un diámetro aerodinámico de 10 µm y 2,5 µm, respectivamente]), que se han asociado en diferentes grados a complicaciones en pacientes con enfermedad de células falciformes, sin una imagen coherente emergente. Existe buena evidencia de que el asma es exacerbada por los contaminantes del aire, particularmente el ozono, y existe una fuerte asociación entre el asma y las complicaciones agudas de la anemia drepanocítica.
El análisis y la interpretación de los efectos climáticos y de la calidad del aire son complicados por las estrechas correlaciones entre los diversos factores y la falta de consistencia en las metodologías y enfoques estadísticos. Además, todos los estudios hasta el momento han examinado las asociaciones a nivel de población. La esperanza es que el aumento del uso de sensores móviles para evaluar la exposición individual conduzcan a resultados más claros.
► Otros factores ambientales
El ambiente doméstico es probablemente un determinante mayor de salud en pacientes con enfermedad de células falciformes, aunque este factor sigue siendo en gran parte inexplorado excepto en algunos estudios que sugieren que la exposición primaria o secundaria al humo del tabaco influye en los resultados clínicos y en las complicaciones de esta enfermedad. Además, la alta altitud se ha relacionado con varias complicaciones en la anemia falciforme, presumiblemente debido a los niveles más bajos de oxígeno.
Sin embargo, la evidencia para esta asociación proviene principalmente de pequeños estudios realizados antes de que la hidroxiurea fuera ampliamente utilizada, y los verdaderos efectos de la altitud son poco claros. El infarto esplénico ocurre en el rasgo de células falciformes, y el secuestro esplénico en pacientes con enfermedad por hemoglobina C. El dolor por vaso-oclusión aguda también parece ser más común en los pacientes que viven a gran altitud.
► Enfermedades infecciosas
La infección es un determinante importante del resultado en pacientes con enfermedad de células falciformes, particularmente en niños de África. La infección es probablemente la causa más importante de muerte prematura entre estos niños. La disfunción esplénica tiene un papel clave en el aumento de la susceptibilidad a las infecciones bacterianas en niños con enfermedad de células falciformes, y las infecciones neumocóccicas y por haemophilus parecen ser importantes tanto en el hemisferio norte como en el austral, sugiriendo que las intervenciones básicas, que incluyen la profilaxis con penicilina y la vacunación, podrían conducir a una mejora sustancial en la sobrevida de los pacientes con enfermedad drepanocítica en los países de ingresos más bajos, tal como dichas intervenciones han hecho en los países de altos ingresos.
La malaria es la otra infección que se cree contribuye ampliamente a la mortalidad excesiva entre los pacientes con enfermedad de células falciformes en África, aunque los datos que apoyan esta creencia son escasos. Estudios en Kenya y Tanzania mostraron que la incidencia de la malaria no aumentó entre los pacientes con enfermedad de células falciformes, pero que el riesgo de muerte fue mayor una vez que el paludismo se desarrolló.
En los países de altos ingresos, la infección también contribuye significativamente a la morbi-mortalidad entre los pacientes con enfermedad de células falciformes, particularmente como una causa de muerte en niños (Streptococcus Pneumoniae) y como causa de osteomielitis (salmonella,
Staphylococcus aureus, bacilos gram negativos, y Mycobacterium tuberculosis) y de síndrome torácico agudo (clamidia, micoplasma y virus) en todos los pacientes, independientemente de la edad. Aunque el espectro de infecciones puede variar a través de los diversos entornos, el efecto se modifica grandemente por la disponibilidad de instalaciones para profilaxis y tratamiento, incluyendo el acceso a los antibióticos y a la transfusión de sangre segura.
► Prevención y manejo
Se han establecido programas de detección premarital, prenatal y neonatal en algunos países de altos ingresos, incluyendo partes del Medio Este y los Estados Unidos, pero más importante, dichos programas se están empezando a desarrollar en áreas con una prevalencia muy alta de enfermedad de células falciformes, incluida la India y algunos países africanos.
El desarrollo de pruebas diagnósticas baratas y confiables con alta sensibilidad y especificidad podría facilitar en gran medida la detección de esta enfermedad en estos países de bajos ingresos, especialmente en las zonas rurales de África subsahariana y la India. Sin embargo, si el diagnóstico no es seguido por intervenciones preventivas y el tratamiento con un agente oral económico para prevenir las complicaciones de la enfermedad aguda, la identificación genotípica es casi sin sentido.
Los resultados clínicos han mejorado gradualmente a lo largo de los años, sobre todo como resultado del tratamiento de sostén y el tratamiento con hidroxiurea. Relativamente pocas intervenciones tienen una base de evidencia fuerte, pero aquellas que sí incluyen la profilaxis con penicilina en niños, la prevención del ACV con el uso del doppler transcraneal y la transfusión de sangre, las transfusiones de sangre regulares para prevenir la progresión del infarto cerebral silencioso, y el uso de la hidroxiurea para prevenir el dolor agudo y el síndrome torácico agudo así como el ACV primario.
Con la creciente evidencia de la seguridad y eficacia de la hidroxiurea tanto en adultos como en niños, su uso está aumentando en los países de ingresos altos y bajos, pero continúa siendo subutilizada. Varias otras terapias de moléculas pequeñas están siendo experimentadas en ensayos clínicos.
Además, se ha presentado a la Administración de Alimentos y Drogas una nueva aplicación para la administración de un tratamiento oral de L-glutamina de grado farmacéutico para reducir la frecuencia del dolor y de las hospitalizaciones entre los pacientes con enfermedad de células falciformes. Además, un informe reciente sobre un estudio multicéntrico, fase 2, aleatorizado, controlado con placebo, doble ciego mostró que un anticuerpo monoclonal inhibidor de la P-selectina redujo la frecuencia del dolor agudo en adultos con anemia drepanocítica. Este es un resultado emocionante, dado que la hidroxiurea sigue siendo el único fármaco eficaz para esta indicación.
El trasplante de células madre hematopoyéticas es potencialmente curativo, aunque su uso está restringido por el alto costo, la toxicidad y la disponibilidad limitada de donantes adecuados. Esto se está volviendo potencialmente más aplicable con el desarrollo de regímenes de acondicionamiento menos tóxicos y el uso de fuentes alternativas de células donantes, aunque la donación de células madre alogénicas puede ser reemplazada por la terapia génica y métodos de edición de genes.
Un reporte de caso reciente que describe el uso de un vector lentiviral con sistema de auto-inactivación para inhibir la polimerización de la HbS como prueba del concepto de remisión clínica completa con corrección de hemólisis y marcadores biológicos de la enfermedad refleja ciertamente el ritmo acelerado de la evolución actual de la terapia génica para la enfermedad de células falciformes. Sin embargo, en vista de los desafíos técnicos, económicos, y éticos, parece muy poco probable que estas nuevas terapias sean ampliamente utilizadas en el corto plazo; en el largo plazo, es probable que los altos costos sigan siendo una barrera importante para su disponibilidad, particularmente en el África subsahariana.
Algunas de las intervenciones actualmente utilizadas para la prevención y el tratamiento de la anemia falciforme en los países de altos ingresos serían rentables y podrían salvar la vida de millones de niños en el África subsahariana si se aplican actualmente. Otras intervenciones, como el doppler transcraneal o la transfusión de sangre, podrían ser mucho más difíciles de aplicar en áreas con alta prevalencia de enfermedad de células falciformes y limitada disponibilidad o acceso a la atención médica.
Una mejor comprensión de los modificadores genéticos es esencial para lograr avances en la terapia génica y en el desarrollo de fármacos. Sin embargo, la identificación de los factores de riesgo no genéticos permitiría el asesoramiento individualizado de los pacientes, lo que podría tener un efecto inmediato en la prevención de las complicaciones clínicas y en la mejora de la calidad de vida de cientos de miles de pacientes en todo el mundo con enfermedad de células falciformes.
► Comentario:
La enfermedad de células falciformes es un cuadro clínico complejo cada vez más frecuente a nivel mundial. Aunque el diagnóstico precoz, las medidas profilácticas y terapéuticas y el trasplante de células madre hematopoyéticas pueden mejorar ampliamente la sobrevida y la calidad de vida de los pacientes con esta enfermedad, la comprensión del rol de los factores genéticos y no genéticos en la diversidad fenotípica de esta enfermedad sigue siendo limitada. La investigación de estos parámetros, junto con el desarrollo de nuevas terapéuticas permitirán mejorar el asesoramiento y el manejo de estos pacientes.
La enfermedad de células falciformes es un cuadro clínico complejo cada vez más frecuente a nivel mundial. Aunque el diagnóstico precoz, las medidas profilácticas y terapéuticas y el trasplante de células madre hematopoyéticas pueden mejorar ampliamente la sobrevida y la calidad de vida de los pacientes con esta enfermedad, la comprensión del rol de los factores genéticos y no genéticos en la diversidad fenotípica de esta enfermedad sigue siendo limitada. La investigación de estos parámetros, junto con el desarrollo de nuevas terapéuticas permitirán mejorar el asesoramiento y el manejo de estos pacientes.
Resumen y comentario objetivo: Dra. María Eugenia Noguerol
Esferocitosis Hereditaria
Esferocitosis Hereditaria
Las anemias hemolíticas asociadas con defectos de membrana eritrocítica pueden ser de dos tipos: hereditarias y adquiridas.Autor: Dr. Checcacci Edgardo*
Desarrollo
Las anemias hemolíticas asociadas con defectos de membrana eritrocítica pueden ser de dos tipos: hereditarias y adquiridas. La esferocitosis hereditaria (EH) es la causa más común de anemia hemolítica en las personas de ascendencia europea, con un predominio de 1 en 5.000.
También puede encontrarse en otros grupos de la población. Se hereda en forma autosómica dominante en el 75% de casos. El 25% restante puede representar mutaciones nuevas u ocasionalmente herencia recesiva o falta de paternidad verdadera.
Se caracterizado por anemia hemolítica de intensidad variable, esferocitos en el extendido de sangre periférica, incremento en la fragilidad osmótica de los eritrocitos y una respuesta clínica favorable a la esplenectomía.
Los eritrocitos de la mayoría de los pacientes con esferocitosis hereditaria muestran algún grado de deficiencia de espectrina, deficiencia que desestabiliza la bicapa lipídica de la membrana. En consecuencia hay un cambio hacia la forma esférica, lo que permite que la célula sea destruida prematuramente en el bazo. El grado de deficiencia de la espectrina se correlaciona con la gravedad clínica del padecimiento, con el grado de fragilidad osmótica y con la respuesta clínica a la esplenectomía.
En sus formas moderadas y en los portadores asintomáticos no se requiere tratamiento; la esplenectomía sigue siendo el tratamiento de elección de la forma típica de esferocitosis hereditaria. La lesión primaria de este padecimiento se encuentra en las proteínas del esqueleto de la membrana y se caracteriza por inestabilidad térmica de la espectrina, desintegración del esqueleto de la membrana después de ser sometida a esfuerzos tensionales, defectos en la interunión de la espectrina y alta susceptibilidad a la digestión por tripsina de la espectrina.
Aspectos Clínicos
Los pacientes pueden diagnosticarse ya en el período del neonatal o en la madurez con o sin los síntomas. Los rasgos cardinales son anemia, ictericia, y esplenomegalia. Algunos pacientes pueden tener todos los síntomas, pero otros son asintomáticos y sólo se descubren por los antecedentes familiares.
La gravedad de la EH es muy variable, pero los recién nacidos afectados con frecuencia presentan enfermedad hemolítica e hiperbilirrubinemia grave. Por otra parte, algunos niños permanecen asintomáticos y se les diagnostica el cuadro porque tienen esplenomegalia o porque se les encuentra anemia leve en un examenn de rutina.
La anemia es la presentación más común (50% de casos); cada uno de los otros rasgos clínicos puede estar presente al diagnóstico en 10% a 15% de casos. Aproximadamente durante el curso de la enfermedad, 50% de pacientes desarrollan la ictericia, y 50% desarrollan un bazo palpable en el primer año de vida. En la adultez, el 95% de pacientes habría desarrollado un bazo palpable.
Aquellos pacientes con formas graves presentan palidez, astenia, ictericia, e intolerancia al ejercicio durante la infancia. Debido al recambio eritrocitario acelerado, los enfermos pueden desarrollar litiasis biliar y son susceptibles a crisis aplásticas. La infección por parvovirus y otros virus pueden inhibir la reticulocitosis y causar una crisis aplástica con anemia aguda muy intensa.
Exámenes de laboratorio:
La anemia muy probablemente es mas severa en la niñez temprana (<1.55 mmol/L [<10 g/dL]) que después en la vida. Los reticulocitos casi invariablemente están elevados, pero la hiperbilirubinemia sólo ocurre aproximadamente en 50% de casos. La concentración de hemoglobina corpuscular media aumenta (>36) en 50% de pacientes que tienen EH, pero la hemoglobina corpuscular media y el volumen corpuscular medio normalmente es normal. El extendido de sangre periférica puede presentarse el esferocito clásico en el 80% de casos. La prueba de diagnóstico definitiva es la prueba de fragilidad osmótica incubada que muestra un modelo de fragilidad aumentada en la EH.
Dificultades en el diagnóstico
También puede encontrarse en otros grupos de la población. Se hereda en forma autosómica dominante en el 75% de casos. El 25% restante puede representar mutaciones nuevas u ocasionalmente herencia recesiva o falta de paternidad verdadera.
Se caracterizado por anemia hemolítica de intensidad variable, esferocitos en el extendido de sangre periférica, incremento en la fragilidad osmótica de los eritrocitos y una respuesta clínica favorable a la esplenectomía.
Los eritrocitos de la mayoría de los pacientes con esferocitosis hereditaria muestran algún grado de deficiencia de espectrina, deficiencia que desestabiliza la bicapa lipídica de la membrana. En consecuencia hay un cambio hacia la forma esférica, lo que permite que la célula sea destruida prematuramente en el bazo. El grado de deficiencia de la espectrina se correlaciona con la gravedad clínica del padecimiento, con el grado de fragilidad osmótica y con la respuesta clínica a la esplenectomía.
En sus formas moderadas y en los portadores asintomáticos no se requiere tratamiento; la esplenectomía sigue siendo el tratamiento de elección de la forma típica de esferocitosis hereditaria. La lesión primaria de este padecimiento se encuentra en las proteínas del esqueleto de la membrana y se caracteriza por inestabilidad térmica de la espectrina, desintegración del esqueleto de la membrana después de ser sometida a esfuerzos tensionales, defectos en la interunión de la espectrina y alta susceptibilidad a la digestión por tripsina de la espectrina.
Aspectos Clínicos
Los pacientes pueden diagnosticarse ya en el período del neonatal o en la madurez con o sin los síntomas. Los rasgos cardinales son anemia, ictericia, y esplenomegalia. Algunos pacientes pueden tener todos los síntomas, pero otros son asintomáticos y sólo se descubren por los antecedentes familiares.
La gravedad de la EH es muy variable, pero los recién nacidos afectados con frecuencia presentan enfermedad hemolítica e hiperbilirrubinemia grave. Por otra parte, algunos niños permanecen asintomáticos y se les diagnostica el cuadro porque tienen esplenomegalia o porque se les encuentra anemia leve en un examenn de rutina.
La anemia es la presentación más común (50% de casos); cada uno de los otros rasgos clínicos puede estar presente al diagnóstico en 10% a 15% de casos. Aproximadamente durante el curso de la enfermedad, 50% de pacientes desarrollan la ictericia, y 50% desarrollan un bazo palpable en el primer año de vida. En la adultez, el 95% de pacientes habría desarrollado un bazo palpable.
Aquellos pacientes con formas graves presentan palidez, astenia, ictericia, e intolerancia al ejercicio durante la infancia. Debido al recambio eritrocitario acelerado, los enfermos pueden desarrollar litiasis biliar y son susceptibles a crisis aplásticas. La infección por parvovirus y otros virus pueden inhibir la reticulocitosis y causar una crisis aplástica con anemia aguda muy intensa.
Exámenes de laboratorio:
La anemia muy probablemente es mas severa en la niñez temprana (<1.55 mmol/L [<10 g/dL]) que después en la vida. Los reticulocitos casi invariablemente están elevados, pero la hiperbilirubinemia sólo ocurre aproximadamente en 50% de casos. La concentración de hemoglobina corpuscular media aumenta (>36) en 50% de pacientes que tienen EH, pero la hemoglobina corpuscular media y el volumen corpuscular medio normalmente es normal. El extendido de sangre periférica puede presentarse el esferocito clásico en el 80% de casos. La prueba de diagnóstico definitiva es la prueba de fragilidad osmótica incubada que muestra un modelo de fragilidad aumentada en la EH.
Dificultades en el diagnóstico
EH Neonatal
La mayoría de los pacientes son sintomático en el período neonatal, normalmente con ictericia que aparece en las primeras 48 horas. De vez en cuando, la ictericia aparece después. La anemia generalmente no es severa a menos que se compique por la anemia fisiológica, y la esplenomegalia es rara. Debido a que la respuesta de la médula ósea a la anemia en el período neonatal no es rápida, particularmente durante el período de anemia fisiológica, la reticulocitosis no es una señal fidedigna. La haptoglobina del suero como un indicador de hemólisis no es fiable en el período del neonatal.
Los glóbulos rojos fetales generalmente son más resistentes a la hemólisis osmótica; por consiguiente, la prueba de fragilidad osmótica no se recomienda, aunque la prueba incubada es fiable. Los sepsis bacterianos pueden simular un cuadro similar al neonatal EH.
EH Leve
Aproximadamente el 20% al 30% de pacientes que hacen EH tienen enfermedad leve y permanecen asintomáticos o sólo muy ligeramente u ocasionalmente sintomáticos hasta la adultez, cuando ellos presentan las complicaciones como los cálculos biliares. La historia puede revelar episodios de ictericia con o sin el dolor del cuadrante superior derecho. Los resultados del examen clínico pueden incluir el esplenomegalia leve, y los estudios hematológicos pueden revelar hemólisis compensado con reticulocitosis mínima y pocos esferocitos. La prueba de fragilidad osmótica incubada normalmente proporciona el diagnóstico. La hemólisis puede agravarse por el embarazo o puede ejercicio, y puede ser familiar o puede aparecer esporádicamente en familias que incluyen a los miembros muy afectados.
EH Severa
Menos del 5% de los pacientes tienen enfermedad severa que probablemente es el resultado de las formas recesivas de la enfermedad. Normalmente se presentan con anemia severa en la infancia y pueden depender de las transfusiones. La esplenomegalia lleva a la resolución de la anemia y de la apariencia de los esferocitos en sangre periférica.
Complicaciones
Aunque la anemia hemolítica crónica es la norma en EH, otras formas de anemia pueden complicar el cuadro. La aplasia de médula ósea inducida por virus sigue a la infección por el parvovirus B19 en cualquier paciente que tiene una enfermedad hematólogica que se caracteriza por la supervivencia acortada de los glóbulos rojos. El virus infecta las células eritroides en la médula y acorta su supervivencia. El virus que es responsable en la niñez del eritema infectioso (quinta enfermedad), también puede causar la fiebre, vómitos y dolor abdominal. El examen físico normalmente revela anemia y una disminución en la ictericia.
El recuento de reticulocitos es bajo y se documenta una disminución concomitante pero leve en las plaquetas y glóbulos blancos. La aplasia normalmente dura 10 a 14 días, durante el cual la hemoglobina puede caer al 50% de sus niveles antes de la recuperación gradual, primero en el recuento de reticulocitos, y luego de la hemoglobina. La azoemia leve y la hiperuricemia pueden ocurrir simultáneamente. El compromiso renal concomitante por el parvovirus B19 a veces lleva a la nefropatía. Como el virus también puede afectar a los fetos y puede llevar al aborto, se exigen las precauciones de aislamiento para proteger a las mujeres embarazadas de los niños hospitalizados. Los niños infectados son contagiosos durante el período de aplasia, pero no cuando el exantema aparece.
Pueden asociarse varias enfermedades virales que se acompañan por el hiperactividad del sistema reticuloendotelial con una hiperhemolisis que se caracteriza por aumento de la anemia, reticulocitosis, e ictericia. Estos episodios normalmente no son severos, y ellos se resuelven espontáneamente.
Los cálculos biliares de bilirrubina pueden ocurrir ya a los 3 años de edad, aunque la incidencia máxima se encuentra en la adolescencia y madurez. La incidencia informada es 5% entre los menores de 10 años de edad, 40% a 50% en el grupo etario de 10 -a 40 años y 55% a 75% entre los mayores de 40 años. Aproximadamente 50% de las piedras no producen ningún síntoma y se descubren el mejor por ecografía.
Otras complicaciones informadas son el resultado de la anemia crónica y de la hiperplasia persistente de la médula ósea. Éstos incluyen el retardo del crecimiento y del desarrollo sexual.
Tratamiento
La mayoría de los pacientes son sintomático en el período neonatal, normalmente con ictericia que aparece en las primeras 48 horas. De vez en cuando, la ictericia aparece después. La anemia generalmente no es severa a menos que se compique por la anemia fisiológica, y la esplenomegalia es rara. Debido a que la respuesta de la médula ósea a la anemia en el período neonatal no es rápida, particularmente durante el período de anemia fisiológica, la reticulocitosis no es una señal fidedigna. La haptoglobina del suero como un indicador de hemólisis no es fiable en el período del neonatal.
Los glóbulos rojos fetales generalmente son más resistentes a la hemólisis osmótica; por consiguiente, la prueba de fragilidad osmótica no se recomienda, aunque la prueba incubada es fiable. Los sepsis bacterianos pueden simular un cuadro similar al neonatal EH.
EH Leve
Aproximadamente el 20% al 30% de pacientes que hacen EH tienen enfermedad leve y permanecen asintomáticos o sólo muy ligeramente u ocasionalmente sintomáticos hasta la adultez, cuando ellos presentan las complicaciones como los cálculos biliares. La historia puede revelar episodios de ictericia con o sin el dolor del cuadrante superior derecho. Los resultados del examen clínico pueden incluir el esplenomegalia leve, y los estudios hematológicos pueden revelar hemólisis compensado con reticulocitosis mínima y pocos esferocitos. La prueba de fragilidad osmótica incubada normalmente proporciona el diagnóstico. La hemólisis puede agravarse por el embarazo o puede ejercicio, y puede ser familiar o puede aparecer esporádicamente en familias que incluyen a los miembros muy afectados.
EH Severa
Menos del 5% de los pacientes tienen enfermedad severa que probablemente es el resultado de las formas recesivas de la enfermedad. Normalmente se presentan con anemia severa en la infancia y pueden depender de las transfusiones. La esplenomegalia lleva a la resolución de la anemia y de la apariencia de los esferocitos en sangre periférica.
Complicaciones
Aunque la anemia hemolítica crónica es la norma en EH, otras formas de anemia pueden complicar el cuadro. La aplasia de médula ósea inducida por virus sigue a la infección por el parvovirus B19 en cualquier paciente que tiene una enfermedad hematólogica que se caracteriza por la supervivencia acortada de los glóbulos rojos. El virus infecta las células eritroides en la médula y acorta su supervivencia. El virus que es responsable en la niñez del eritema infectioso (quinta enfermedad), también puede causar la fiebre, vómitos y dolor abdominal. El examen físico normalmente revela anemia y una disminución en la ictericia.
El recuento de reticulocitos es bajo y se documenta una disminución concomitante pero leve en las plaquetas y glóbulos blancos. La aplasia normalmente dura 10 a 14 días, durante el cual la hemoglobina puede caer al 50% de sus niveles antes de la recuperación gradual, primero en el recuento de reticulocitos, y luego de la hemoglobina. La azoemia leve y la hiperuricemia pueden ocurrir simultáneamente. El compromiso renal concomitante por el parvovirus B19 a veces lleva a la nefropatía. Como el virus también puede afectar a los fetos y puede llevar al aborto, se exigen las precauciones de aislamiento para proteger a las mujeres embarazadas de los niños hospitalizados. Los niños infectados son contagiosos durante el período de aplasia, pero no cuando el exantema aparece.
Pueden asociarse varias enfermedades virales que se acompañan por el hiperactividad del sistema reticuloendotelial con una hiperhemolisis que se caracteriza por aumento de la anemia, reticulocitosis, e ictericia. Estos episodios normalmente no son severos, y ellos se resuelven espontáneamente.
Los cálculos biliares de bilirrubina pueden ocurrir ya a los 3 años de edad, aunque la incidencia máxima se encuentra en la adolescencia y madurez. La incidencia informada es 5% entre los menores de 10 años de edad, 40% a 50% en el grupo etario de 10 -a 40 años y 55% a 75% entre los mayores de 40 años. Aproximadamente 50% de las piedras no producen ningún síntoma y se descubren el mejor por ecografía.
Otras complicaciones informadas son el resultado de la anemia crónica y de la hiperplasia persistente de la médula ósea. Éstos incluyen el retardo del crecimiento y del desarrollo sexual.
Tratamiento
Durante los primeros 6 años de vida, si el paciente ha compensado la anemia, está creciendo bien, y puede realizar la mayoría de las actividades que realizan sus pares, es prudente limitar la intervención a 1 mg/d de suplemento de ácido fólico. Como consecuencia, y dependiendo de la severidad de la enfermedad, y como la mayor parte de los esferocitos se destruyen en el bazo, la mayor parte de la hemolisis se elimina mediante la esplenectomía .
El beneficio primario de la esplenectomía que le permite al hematíe tener una vida media cercana a lo normal puede eliminar las complicaciones como la enfermedad de la vesícula biliar. se aconseja efectuarla si no se puede mantener un nivel de hemoglobina de 10 g/dl o un porcentaje de reticulocitos de menor del 10% o si ha habido una crisis aplástica. Se pueden indicar transfusiones a los menores de dos años con enfermedad grave.
El riesgo de sepsis postesplenectomía se disminuye después de los 5 años de edad y puede reducirse más allá administrando vacuna anti-pneumococcical, anti- H. influenzae y anti- meningococcica, idealmente por lo menos 2 semanas antes de la cirugía. Si se inmunizan los pacientes afectados antes de los 2 años de edad, la inmunización debe repetirse después de los 2 años de edad. Otros organismos responsable de la sepsis post esplenectomía incluyen a Escherichia coli y estafilococos. Después de la esplenectomía es necesario administrar indefinidamente penicilina. Las alternativas incluyen eritromicina o amoxicilina
El desarrollo de cálculos biliares puede ser una indicación para la esplenectomía. El tratamiento de los cálculos biliares sintomáticos es la colecistectomía electiva, pero la colelitiasis asintomática puede ser manejada solamente con la observación o mediante colecistectomía electiva.
La respuesta a la esplecnectomía en la EH es del 100% excepto en la presencia de bazos adicionales. Aunque los esferocitos persisten en la circulación, estos tienen una vida media cercana a lo normal en la ausencia del bazo. Los cambios postesplenectomía incluyen el aumento de la hemoglobina y disminución de los reticulocitos.
El beneficio primario de la esplenectomía que le permite al hematíe tener una vida media cercana a lo normal puede eliminar las complicaciones como la enfermedad de la vesícula biliar. se aconseja efectuarla si no se puede mantener un nivel de hemoglobina de 10 g/dl o un porcentaje de reticulocitos de menor del 10% o si ha habido una crisis aplástica. Se pueden indicar transfusiones a los menores de dos años con enfermedad grave.
El riesgo de sepsis postesplenectomía se disminuye después de los 5 años de edad y puede reducirse más allá administrando vacuna anti-pneumococcical, anti- H. influenzae y anti- meningococcica, idealmente por lo menos 2 semanas antes de la cirugía. Si se inmunizan los pacientes afectados antes de los 2 años de edad, la inmunización debe repetirse después de los 2 años de edad. Otros organismos responsable de la sepsis post esplenectomía incluyen a Escherichia coli y estafilococos. Después de la esplenectomía es necesario administrar indefinidamente penicilina. Las alternativas incluyen eritromicina o amoxicilina
El desarrollo de cálculos biliares puede ser una indicación para la esplenectomía. El tratamiento de los cálculos biliares sintomáticos es la colecistectomía electiva, pero la colelitiasis asintomática puede ser manejada solamente con la observación o mediante colecistectomía electiva.
La respuesta a la esplecnectomía en la EH es del 100% excepto en la presencia de bazos adicionales. Aunque los esferocitos persisten en la circulación, estos tienen una vida media cercana a lo normal en la ausencia del bazo. Los cambios postesplenectomía incluyen el aumento de la hemoglobina y disminución de los reticulocitos.
lunes, 12 de noviembre de 2018
Microangiopatía trombótica
Microangiopatía trombótica
Patogenia, manejo y resultados de los síndromes de microangiopatía trombótica.Autor: James N. George, Carla M. Nester N Engl J Med 2014; 371: 654-66
Desarrollo
Los síndromes de microangiopatía trombótica (MAT) son muy diversos. Pueden ser hereditarios o adquiridos. Se producen en niños y adultos. El inicio puede ser repentino o gradual. A pesar de su diversidad, los síndromes MAT están unidos por características clínicas y patológicas definitorias comunes. Las características clínicas incluyen anemia hemolítica microangiopática, trombocitopenia, y daño orgánico. Las características patológicas son el daño vascular que se manifiesta por trombosis arteriolar y capilar con anormalidades características en el endotelio y la pared vascular.
Los autores se centran en nueve trastornos que se describen como síndromes MAT primarios, para los cuales hay evidencia que apoya una anomalía definida como causa probable (Tabla 1). Para mayor claridad en esta discusión, los nombres que se eligieron para estos síndromes reflejan su causa. Sin embargo, se retienen los nombres comunes de púrpura trombocitopénica trombótica (PTT) para la MAT mediada por deficiencia de ADAMTS13 y de síndrome urémico-hemolítico para la MAT mediada por Toxina Shiga (SUH-TS), ya que estos nombres son familiares.
Los autores no utilizan el término "SUH atípico", que fue históricamente utilizado para distinguir ciertos síndromes heterogéneos, no caracterizados, del SUH-TS, ya que el término carece de especificidad y de sugerencia de causa. Asimismo, no se utiliza el término "idiopático" para ninguno de los síndromes MAT primarios.
La presencia de una anomalía causal, como la deficiencia de ADAMTS13 o una mutación del complemento, puede no expresarse clínicamente hasta que una condición, como el embarazo, una cirugía, o un trastorno inflamatorio, precipita un episodio agudo de MAT. El tratamiento de estos pacientes se centra en la causa del síndrome MAT primario, no en la condición precipitante. Estos pacientes son distintos de muchos otros pacientes que tienen anemia hemolítica microangiopática y trombocitopenia como manifestaciones de un trastorno subyacente (Tabla 2). El tratamiento de estos pacientes se centra en el trastorno subyacente.
El conocimiento de la patogenia, el manejo y los resultados de los síndromes MAT primarios se ha acelerado en los últimos años. El objetivo de esta revisión es proporcionar una perspectiva unificada de estos síndromes.
*Los enumerados son trastornos que pueden sugerir inicialmente el diagnóstico de un síndrome MAT primario. Muchas infecciones sistémicas diferentes (virales, tales como el virus de la inmunodeficiencia humana y citomegalovirus; hongos, tales como Aspergillus; y bacterianas) y muchos diferentes tipos de cáncer sistémicos pueden estar asociados con anemia hemolítica microangiopática y trombocitopenia sin coagulación intravascular diseminada (CID) manifiesta. Muchos otros trastornos, tales como cualquier condición asociada con CID, también pueden presentarse con anemia hemolítica microangiopática y trombocitopenia y también deben ser considerados como causas alternativas en la evaluación de los pacientes con un posible diagnóstico de síndrome MAT primario. Algunos de estos trastornos (por ejemplo, hipertensión grave, lupus eritematoso sistémico, y esclerosis sistémica) también pueden estar asociados con las características patológicas de MAT. Estos trastornos pueden causar directamente las características clínicas y patológicas de la MAT, una hipótesis apoyada por la resolución de estas características con el tratamiento eficaz del trastorno. HELLP denota hemólisis, niveles elevados de enzimas hepáticas y plaquetas bajas.
1. PTT (Adquirida y Hereditaria)
Antecedentes
En 1924, Moschcowitz describió a una niña de 16 años de edad con debilidad, palidez, púrpura, y hemiparesia quien murió después de 14 días por insuficiencia cardíaca. La autopsia reveló trombos hialinos en las arteriolas y los capilares terminales a lo largo de la mayoría de los órganos, incluyendo los riñones. Este informe fue la primera descripción de una MAT, presumiblemente una PTT, también llamada MAT mediada por deficiencia de ADAMTS13.
Causa
En 1982, se observaron multímeros de factor de von Willebrand inusualmente grandes en pacientes con PTT (hereditaria) crónica, en recaída. Este hallazgo condujo al descubrimiento de una proteasa de escisión del factor de von Willebrand, que se caracterizó posteriormente como ADAMTS13. La ADAMTS13 cliva multímeros del factor von Willebrand que son secretados desde las células endoteliales vasculares. La deficiencia de ADAMTS13 resulta en multímeros del factor de von Willebrand inusualmente grandes, con riesgo de trombos plaquetarios en vasos de pequeño calibre.
La PTT hereditaria (también llamada síndrome de Upshaw-Schulman) es causada por mutaciones homocigotas o heterocigotas compuestas de la ADAMTS13. Los pacientes con mutaciones heterocigotas no tienen anormalidades aparentes. La PTT adquirida es una enfermedad autoinmune causada por la inhibición por autoanticuerpos de la actividad de la ADAMTS13. La incidencia de PTT adquirida es mucho mayor en adultos (2,9 casos por 1 millón por año) que en niños (0,1 casos por 1 millón por año). Los factores que están asociados con una mayor frecuencia de este trastorno incluyen la edad entre 18 y 50 años, la raza negra, y el sexo femenino.
Cuadro clínico y diagnóstico Entre los síndromes MAT principales, la PTT es única porque rara vez causa daño renal agudo severo. Las características clínicas de la PTT hereditaria son episodios recurrentes de anemia hemolítica microangiopática y trombocitopenia, a menudo con alteraciones neurológicas u otros signos de lesión orgánica. El diagnóstico de PTT hereditaria requiere la documentación de la deficiencia de ADAMTS13 y la ausencia de un autoanticuerpo inhibidor de la misma, y la confirmación requiere la documentación de mutaciones de la ADAMTS13.
La PTT hereditaria puede ser evidente al nacimiento, con anemia hemolítica microangiopática y trombocitopenia, o no ser evidente hasta la edad adulta, cuando puede ser precipitada por una condición como el embarazo. Aunque la severidad de la condición puede estar relacionada con mutaciones de ADAMTS13, las observaciones de heterogeneidad entre hermanos sugieren que las manifestaciones clínicas requieren de factores genéticos o ambientales adicionales, similares a las observaciones en ratones deficientes de ADAMTS13.
Las características clínicas presentes en la PTT adquirida son diversas; algunos pacientes tienen alteraciones mínimas, mientras que otros están críticamente enfermos. Son comunes la debilidad, los síntomas gastrointestinales, la púrpura, y las alteraciones neurológicas focales transitorias. Sin embargo, un tercio de los pacientes no tienen anormalidades neurológicas. La mayoría de los pacientes tienen valores de creatinina normales o ligeramente elevados sólo de manera transitoria.
Los criterios de diagnóstico son la presencia de anemia hemolítica microangiopática y de trombocitopenia sin otra causa aparente. Por lo tanto, a veces no es posible la exclusión de otros síndromes MAT primarios. Un nivel de ADAMTS13 que indica menos del 10% de actividad normal apoya el diagnóstico clínico de PTT adquirida. Identifica casi todos los pacientes con riesgo de recaída, pero este nivel no es ni suficientemente sensible para identificar a todos los pacientes con PTT ni suficientemente específico para excluir a los pacientes con trastornos subyacentes.
Tratamiento
El tratamiento para la PTT hereditaria es el reemplazo de la ADAMTS13 mediante infusión de plasma. Los pacientes con reacciones alérgicas graves al plasma han sido efectivamente tratados con concentrado de factor VIII derivado de plasma que contiene ADAMTS13. Aunque muchos pacientes requieren plasma sólo cuando se producen trombocitopenia o síntomas, otros pueden requerir infusiones de plasma profilácticas en forma regular.
Antes del uso de la plasmaféresis, la sobrevida de los pacientes con PTT adquirida era del 10%. En 1991, un estudio aleatorizado controlado documentó una tasa de sobrevida del 78% con la plasmaféresis. La alta mortalidad sin tratamiento crea la urgencia de empezar el intercambio de plasma, que a menudo lleva a tratar a pacientes que no tienen PTT. Los glucocorticoides son el tratamiento estándar; el rituximab y otros agentes inmunosupresores son apropiados cuando el curso clínico es complicado. Raramente se requiere diálisis.
Resultados a largo plazo
Los resultados a largo plazo de los pacientes con PTT hereditaria son desconocidos. Los datos experimentales sugieren que la ADAMTS13 proporciona protección contra la aterosclerosis, pero no se sabe si los pacientes con PTT hereditaria tienen un mayor riesgo de enfermedad cardiovascular. El seguimiento a largo plazo de los pacientes con PTT adquirida puso de manifiesto el riesgo de recaída y un aumento de la prevalencia de deterioro cognitivo, depresión mayor, lupus eritematoso sistémico, hipertensión y muerte.
Necesidades futuras
Si el seguimiento a largo plazo muestra que la PTT hereditaria causa un aumento de la morbilidad, el tratamiento profiláctico será el punto más importante. El desarrollo de ADAMTS13 recombinante haría que el tratamiento profiláctico sea más simple y más seguro. Para el tratamiento de pacientes con PTT adquirida, se requieren alternativas más seguras y más accesibles que la plasmaféresis.
2. MAT mediada por Complemento (Adquirida y Hereditaria)
Antecedentes
Esta MAT que se caracteriza predominantemente por falla renal y se describe como SUH fue reconocida como un trastorno familiar en 1975. En 1981, se halló que dos hermanos con MAT tenían una deficiencia del factor H del complemento. La asociación entre la MAT y mutaciones en el gen que codifica el factor H del complemento (FHC) se estableció en 1998. Posteriormente, se identificaron mutaciones en múltiples otros factores que facilitan el aumento de la activación de la vía alternativa del complemento en pacientes con MAT.
Causa
La MAT mediada por complemento resulta de la activación incontrolada de la vía alternativa del complemento. A diferencia de las otras dos vías de activación del complemento, la vía alternativa es constitutivamente activa como resultado de la hidrólisis espontánea de C3 a C3b.
En ausencia de una regulación normal, la deposición de C3b sobre los tejidos puede aumentar notablemente, lo que resulta en aumento de la formación del complejo terminal C5b-9 del complemento (también llamado complejo de ataque de membrana) y lesión de las células normales. El papel preciso de la desregulación del complemento en la MAT no ha sido plenamente definido. Puede estar involucrada la lesión endotelial así como la desregulación del complemento en la superficie de las plaquetas causando activación.
La MAT mediada por complemento hereditaria puede resultar de una mutación con pérdida de función de un gen regulador (FHC, CFI, o CD46) o de una mutación con ganancia de función en un gen efector (CFB o C3). La mayoría de las mutaciones del complemento que se asocian con MAT son heterocigotas, incluso aunque muchos miembros de la familia con mutaciones heterocigotas sean asintomáticos.
Puede explicar esta discrepancia una diferencia entre probandos y miembros de la familia en la presencia de genes modificadores adicionales. Fueron identificadas otras anormalidades genéticas en pacientes con MAT mediada por complemento, incluyendo polimorfismos de un único nucleótido en FHC y CD46, variaciones del número de copias en los genes 1 y 3 relacionados con el FHC (FHCR1 y FHCR3), y fusión de genes de la región FHCR con FHC causado por recombinación homóloga no alélica. Estas anomalías genéticas adicionales pueden contribuir a la pérdida de regulación de la vía alternativa y al aumento del riesgo de MAT.
Además de las anomalías genéticas, una deficiencia funcional en el factor H del complemento puede resultar de anticuerpos para el complemento, dando lugar a una MAT adquirida. Estos anticuerpos FHC se corresponden con aproximadamente el 10% de las MAT mediadas por complemento. Estos anticuerpos son responsables de la protección celular defectuosa dependiente del FHC.
Cuadro clínico y diagnóstico
La lesión renal aguda y la hipertensión son alteraciones prominentes en la MAT mediada por complemento. Los criterios diagnósticos actuales son aquellos que se utilizaron en ensayos clínicos con un total de 37 pacientes, y que apoyaron la aprobación de eculizumab (un anticuerpo monoclonal humanizado que bloquea la generación de C5a y C5b) para el tratamiento del "SUH atípico" en 2011.
Estos criterios incluyen todo lo siguiente: un nivel de creatinina sérica en o por encima del límite superior del rango normal, anemia hemolítica microangiopática, trombocitopenia, actividad de ADAMTS13 del 5% o más, y análisis de materia fecal negativo para infección por germen productor de toxina Shiga.
Estos criterios no son específicos; también pueden ocurrir en todos los otros síndromes MAT primarios, así como en otros pacientes con anemia hemolítica microangiopática y trombocitopenia. Los estudios genéticos del complemento, ahora disponibles en el mercado con resultados rápidos, pueden proporcionar un diagnóstico más específico. Los niveles plasmáticos normales de C3, C4, y factores de complemento H, B, e I no excluyen el diagnóstico de MAT mediada por complemento.
Tratamiento
Puede utilizarse terapia anti-complemento para complementar el tratamiento con plasma y adelantarse a un potencial trasplante hepático. El eculizumab es actualmente el único agente anti-complemento disponible. Su efecto puede ser limitado en los pacientes que tienen mutaciones de C5. Los criterios diagnósticos no específicos y el hecho de que los pacientes con mutaciones del complemento no identificadas pueden tener una respuesta a la terapia anti-complemento hacen que sea difícil tomar la decisión de utilizar un agente anti-complemento como terapia inicial.
La terapia anti-complemento es un tratamiento inicial razonable para los pacientes con anticuerpos contra el factor H del complemento. Sin embargo, debe considerarse el uso de la inmunosupresión para reducir el título de anticuerpos. El alto costo del eculizumab (costo de adquisición al por mayor de 1 año de tratamiento para un adulto, $ 614,736; Farmacia del Centro Médico de la Universidad de Oklahoma, 5 de marzo de 2014) y que debe continuarse indefinidamente son temas críticos. Debe considerarse el riesgo de infección meningocóccica en relación con el tratamiento con eculizumab.
Resultados a largo plazo
Antes del uso de eculizumab, los riesgos de insuficiencia renal en fase terminal o muerte y de recurrencia después de un trasplante de riñón eran principalmente dependientes del análisis mutacional, con las mutaciones del FHC causando el mayor riesgo. Se supone que el tratamiento anti-complemento mejorará los resultados.
Necesidades futuras Son necesarios ensayos clínicos para determinar la duración más adecuada de la terapia anti-complemento y para desarrollar marcadores de vigilancia para confirmar la remisión renal. Se requiere más investigación genética para explicar la heterocigosidad y la baja penetrancia de las mutaciones del complemento actualmente identificadas y también para determinar la causa de enfermedad en pacientes que pueden tener MAT mediada por complemento, pero en quienes no se ha identificado una mutación.
3. Síndrome Urémico Hemolítico mediado por Toxina Shiga
Antecedentes
El nombre "Síndrome Urémico Hemolítico" se propuso por primera vez en 1955. Las características clínicas típicas del SUH-TS fueron descriptas por primera vez en un reporte de 1962 que implicó a cinco niños de 6 a 10 meses de edad que habían tenido diarrea previa a la falla renal. Se sospechó una causa infecciosa, pero la asociación entre SUH-TS e infección entérica, Shigella dysenteriae tipo 1, no se reconoció hasta 1975. No fue sino hasta 1983, durante una investigación de brotes de colitis hemorrágica, que la Escherichia coli O157:H7 fue identificada como un patógeno.
El mismo año, se describió la asociación entre SUH-TS y E. coli productora de toxina Shiga. Múltiples cepas de E. coli producen toxina Shiga; la E. coli O157:H7 es el patógeno más común asociado con SUH-TS en Europa y América. S. dysenteriae tipo 1 sigue siendo una causa endémica de SUH-TS en otros países. Aunque el SUH-TS se popularizó por sus grandes brotes, la mayoría de las ocurrencias son esporádicas. El SUH-TS es mucho más común entre los niños (edad media, 2 años), en los cuales la mortalidad es del 3%. El SUH-TS en adultos es más grave, con mayor mortalidad.
Causa
E. coli productora de toxina Shiga es una bacteria intestinal común en el ganado, en consonancia con el predominio rural del SUH-TS endémico. Los brotes provienen de productos contaminados, como agua, carne vacuna, verduras y otros alimentos. El daño celular se produce cuando la toxina Shiga se une a la globotriaosilceramida (Gb3, también conocida como CD77 o trihexósido de ceramida) de las células endoteliales, así como a las células mesangiales y epiteliales renales (podocitos y células tubulares).
La apoptosis celular resulta de la unión a Gb3, endocitosis, transporte retrógrado, y translocación citosólica de la toxina Shiga, con la subsiguiente inactivación ribosomal. La toxina Shiga también es activadora celular, pro-inflamatoria, y pro-trombótica y facilita la trombosis mediante la inducción de la secreción de factor von Willebrand.
Cuadro clínico y diagnóstico
Se manifiesta por dolor abdominal intenso y diarrea, a menudo sanguinolenta, varios días después de consumir alimentos contaminados. La trombocitopenia y el fallo renal comienzan con la resolución de los síntomas gastrointestinales. La toxina Shiga se identifica mediante análisis de materia fecal durante la fase de diarrea aguda, pero puede no ser identificable cuando comienza el SUH-TS.
Tratamiento
El tratamiento sigue siendo de sostén. La hidratación temprana, agresiva, tiene un rol de protección renal. Los pacientes comúnmente requieren diálisis. Los beneficios de la plasmaféresis y del tratamiento anti-complemento son inciertos.
Resultados a largo plazo
La hipertensión y las anomalías neurológicas pueden persistir después de que resuelva la fase aguda. Rara vez ocurre enfermedad renal en fase terminal.
Necesidades futuras
La mayor necesidad es la prevención de las infecciones enterohemorrágicas y del SUH-TS mediante medidas de salud pública (por ejemplo, seguridad e higiene de los alimentos). Nuevos agentes que neutralizan la toxina Shiga pueden proporcionar un tratamiento eficaz para el SUH-TS.
4. MAT mediada por Drogas (Reacción Inmune)
Antecedentes
Las reacciones adversas a drogas pueden ser causadas por reacciones inmunológicas, idiosincrásicas, no relacionadas con la dosis o por efectos tóxicos que son dependientes de la dosis y el tiempo. Ambos tipos de reacciones se asocian con MAT. La reacción inmune en la MAT mediada por fármacos fue reconocida por primera vez en 1980 en un paciente que había repetido episodios de insuficiencia renal aguda, hemólisis y trombocitopenia después de exposiciones documentadas a quinina.
Estudios posteriores confirmaron el patrón de recurrencia e implicaron más a la quinina al describir anticuerpos dependientes de quinina que eran reactivos con múltiples tipos celulares. Aunque se asociaron muchas otras drogas con MAT, sólo la MAT asociada a quinina ha sido apoyada por la documentación de anticuerpos fármaco-dependientes. Quetiapina y gemcitabina son las otras drogas para las que la asociación con episodios agudos de MAT ha sido apoyada por episodios agudos recurrentes ante exposiciones repetidas.
Causa
La unión del anticuerpo fármaco-dependiente a los antígenos de múltiples células puede ser facilitada por elementos estructurales que contienen los fármacos y que son complementarios a los dominios tanto del epítope como del anticuerpo. Los anticuerpos dependientes de quinina pueden mediar la MAT, en parte, por la activación de células endoteliales.
Cuadro clínico y diagnóstico
Se puede sospechar MAT mediada por drogas ante la aparición repentina de síntomas sistémicos graves, a menudo con lesión renal aguda anúrica, en cuestión de horas después de la exposición al fármaco. Puede haber un historial de enfermedad tras exposiciones previas al fármaco sospechoso. La asociación con la quinina puede ser pasada por alto porque la exposición puede ocurrir a lo largo de los años y puede no ser reportada por el paciente sin preguntas explícitas. La exposición puede incluir comprimidos o bebidas que contienen quinina. La documentación de anticuerpos fármaco-dependientes apoya el diagnóstico clínico; sin embargo, una prueba negativa no excluye la asociación con drogas.
Tratamiento
El tratamiento de sostén y la evitación del fármaco pueden ser las únicas medidas beneficiosas. Es frecuente que se inicie la plasmaféresis por la sospecha de PTT y porque la causa mediada por fármacos es incierta.
Resultados a largo plazo
Es frecuente la enfermedad renal crónica con hipertensión. Puede ocurrir enfermedad renal en etapa terminal.
Necesidades Futuras
Se requieren pruebas para la detección de anticuerpos fármaco-dependientes rápidamente disponibles para ayudar en el diagnóstico clínico. Entender el mecanismo de lesión renal aguda puede dar una idea para el tratamiento objetivo.
5. MAT mediada por Drogas (Reacción Tóxica Relacionada con la Dosis)
Antecedentes
Muchos fármacos, incluyendo agentes inmunosupresores y quimioterapéuticos e inhibidores del factor de crecimiento endotelial vascular (FCEV), fueron reportados como causa de MAT mediante toxicidad dosis y tiempo dependiente. La evidencia que apoya una relación causal es limitada.
Causa Puede haber múltiples mecanismos de lesión renal mediada por drogas tóxicas. Entre los probables roles de los inhibidores de la calcineurina (como la ciclosporina y el tacrolimus) está su capacidad de causar disfunción endotelial y aumento de la agregación plaquetaria, posiblemente a través de la inhibición de la prostaciclina. La inhibición de la función del FCEV en las células endoteliales y los podocitos renales causa el desarrollo gradual de MAT glomerular.
Cuadro clínico y diagnóstico
La presentación típica es la pérdida gradual de la función renal con hipertensión. Se puede producir MAT abrupta, severa, al igual que con un mal uso intravenoso del opiáceo oximorfona.
Tratamiento
El tratamiento de apoyo y la evitación del fármaco pueden ser el único manejo beneficioso. Para algunos medicamentos, tales como los inhibidores de la calcineurina, la reducción de la dosis, más que la evitación del fármaco, puede ser suficiente.
Resultados a largo plazo
La anemia hemolítica microangiopática y la trombocitopenia suelen resolver. La insuficiencia renal puede persistir.
Necesidades futuras
Es esencial determinar los fármacos que provocan MAT, o identificar explicaciones alternativas para la MAT en estos pacientes.
6. MAT mediada por Metabolismo
Antecedentes
La Enfermedad de la Cobalamina C es un trastorno hereditario del metabolismo de la cobalamina (vitamina B12) que puede causar MAT y disfunción orgánica múltiple en los lactantes. Además, se ha reportado MAT en un adulto.
Causa
Los trastornos del metabolismo de la cobalamina resultan de mutaciones homocigotas o heterocigotas compuestas en un gen que codifica la proteína tipo C de la aciduria metilmalónica y la homocistinuria (MMACHC). La deficiencia resultante de metilcobalamina causa hiperhomocisteinemia, disminución de los niveles plasmáticos de metionina, y aciduria metilmalónica. El metabolismo anormal de la cobalamina C se asocia con activación plaquetaria, generación de especies de oxígeno reactivo, disfunción endotelial, expresión aumentada del factor tisular, y activación de la coagulación.
Cuadro clínico y diagnóstico
Los lactantes con enfermedad de la cobalamina C tienen diversas anomalías del desarrollo. El único adulto reportado se presentó con anemia hemolítica microangiopática, trombocitopenia, insuficiencia renal aguda, e hipertensión; no tenía antecedentes personales o familiares relevantes.
Tratamiento
La hidroxicobalamina parenteral es el principal tratamiento para los niños. Cuando el paciente adulto antes mencionado no tuvo una respuesta al tratamiento anti-complemento, se sospechó un defecto intracelular del metabolismo de la vitamina B12 y se confirmó mediante pruebas de rutina disponibles, que mostraron hiperhomocisteinemia, disminución de metionina en plasma, aciduria metilmalónica, y niveles plasmáticos normales de vitamina B12. El paciente respondió al tratamiento con hidroxicobalamina, betaína, y ácido folínico.
Resultados a largo plazo
Las secuelas neurológicas son comunes en los lactantes afectados. Se reportaron enfermedad renal crónica con hipertensión y proteinuria hasta en un 40% de los pacientes. La enfermedad renal en etapa terminal y la recurrencia de MAT parecen prevenirse con el tratamiento de reemplazo.
Necesidades futuras
Se requiere mayor conciencia de la MAT inducida por cobalamina C y un mayor uso de las pruebas de detección de rutina para la deficiencia de cobalamina.
7. MAT mediada por Factores de la Coagulación
Antecedentes
Se identificaron anomalías genéticas de la trombomodulina, el plasminógeno, y una proteína asociada a la proteína quinasa C, diacilglicerol quinasa ε (DGQE), en pacientes con MAT. Estos hallazgos sugieren que puede haber un papel primordial para las proteínas de la coagulación en la patogénesis de los Síndromes MAT.
Causa
La cuestión de si el papel de la trombomodulina en la MAT está principalmente relacionado con la coagulación o si está mediada únicamente por el complemento requiere mayor estudio. Se documentó el rol de la DGQE en dos informes que describen 22 pacientes en 12 familias. Todos los pacientes tenían mutaciones homocigotas o heterocigotas compuestas de la DGQE. Los pacientes con mutaciones heterocigotas no tenían anomalías clínicas. En un informe no se identificaron mutaciones de genes reguladores del complemento.
Se propuso que una mutación homocigota en el gen que codifica la DGQE conduce a una pérdida de función, resultando en activación de la proteína quinasa C (PQC). La activación de la PQC facilita la sobre regulación de factores protrombóticos (factor de von Willebrand, factor tisular, e inhibidor del activador del plasminógeno tipo 1) y la baja regulación del receptor del FCEV.
La disminución de la función del FCEV con la consiguiente lesión de podocitos renales también puede ser una consecuencia de mutaciones de la DGQE. La suma de estos eventos es un cambio hacia un estado protrombótico. La comunicación entre el sistema de la coagulación y el sistema del complemento puede tener un papel en la patogénesis de la MAT.
Cuadro clínico y diagnóstico
Los pacientes con mutaciones DGQE se presentan con lesión renal aguda. La mayoría de estos pacientes estudiados eran menores de 1 año.
Tratamiento Los beneficios de la infusión o el intercambio de plasma y la inmunosupresión fueron inconsistentes. Hubo recaídas mientras los pacientes estaban recibiendo terapia anti-complemento.
Resultados a largo plazo
Es común la enfermedad renal en etapa terminal. Cuatro niños con este trastorno fueron sometidos a trasplante de riñón; uno tuvo injuria renal recurrente.
Necesidades futuras
Es esencial la determinación de la prevalencia de estos síndromes y la comprensión de los mecanismos que conducen a la MAT.
Mecanismos de lesión orgánica
Las causas antes mencionadas de síndromes MAT primarios no explican por qué la lesión renal es predominante en todos salvo en la PTT. La PTT causa trombos en la extensa microvasculatura renal, aunque la lesión renal aguda severa y la enfermedad renal crónica son raras. La diferencia puede ser que la patogénesis de la PTT se relaciona principalmente con trombosis vascular, mientras que la patogénesis de los otros síndromes MAT primarios también incluye daño a las células renales y células endoteliales residentes.
Por ejemplo, en pacientes con SUH-TS, la toxina Shiga causa daño a los podocitos epiteliales, células mesangiales, y células tubulares renales. Los inhibidores del FCEV afectan a los podocitos epiteliales glomerulares, y la mayor activación de la proteína quinasa C en pacientes con mutaciones de la DGQE puede causar injuria en los podocitos. Comprender el mecanismo de lesión en células renales residentes en la MAT mediada por complemento, por anticuerpos quinina-dependientes, o en la Enfermedad de la Cobalamina C requiere más estudio.
También hay claras diferencias con respecto al riesgo de enfermedad renal crónica entre los siete síndromes MAT primarios en los que la lesión renal aguda está normalmente presente. Entre los síndromes adquiridos, rara vez se desarrolla lesión renal crónica en pacientes con SUH-TS, tal vez porque el SUH-TS no recurre.
Sin embargo, los pacientes con un solo episodio reconocido de MAT inducido por quinina comúnmente tienen lesión renal crónica. Entre los síndromes hereditarios, también hay una variedad sustancial en la gravedad de la lesión renal.
Los pacientes con mutaciones del complemento pueden presentar lesión renal aguda a cualquier edad, mientras que aquellos con mutaciones DGQE suelen presentar lesión renal aguda en la infancia. Estas variaciones en las manifestaciones clínicas de los síndromes MAT enfatizan la necesidad de una mayor comprensión de los mecanismos de la enfermedad.
Conclusiones
En los últimos años, ha habido una aceleración dramática en la comprensión de los síndromes MAT primarios. Los nuevos descubrimientos con respecto a los mecanismos causales crearon oportunidades para tratamientos específicos. La disponibilidad de tratamientos específicos llevó a la necesidad de diagnósticos rápidos y precisos. El uso de tratamientos eficaces ha disminuido la mortalidad, pero también ha puesto de manifiesto morbilidades a largo plazo anteriormente no reconocidas. Estos avances predicen la aceleración continua de la comprensión de los principales síndromes MAT.
Comentario: Los síndromes de microangiopatía trombótica son un conjunto heterogéneo de trastornos hereditarios o adquiridos, cuyas características clínicas incluyen anemia hemolítica microangiopática, trombocitopenia, y daño orgánico variable. La presencia de una anomalía causal puede no expresarse clínicamente hasta que cierta condición (como embarazo, cirugía, o proceso inflamatorio) precipita el episodio agudo de MAT, y el tratamiento se centra básicamente en la misma.
Si bien han aumentado notablemente los conocimientos con respecto a los síndromes MAT primarios, se requieren nuevas investigaciones que ayuden a la comprensión de estos trastornos, a fin de mejorar las opciones de tratamiento y disminuir la morbi-mortalidad que surge de los mismos.
Resumen y comentario objetivo: Dra. María Eugenia Noguerol
Los autores se centran en nueve trastornos que se describen como síndromes MAT primarios, para los cuales hay evidencia que apoya una anomalía definida como causa probable (Tabla 1). Para mayor claridad en esta discusión, los nombres que se eligieron para estos síndromes reflejan su causa. Sin embargo, se retienen los nombres comunes de púrpura trombocitopénica trombótica (PTT) para la MAT mediada por deficiencia de ADAMTS13 y de síndrome urémico-hemolítico para la MAT mediada por Toxina Shiga (SUH-TS), ya que estos nombres son familiares.
Los autores no utilizan el término "SUH atípico", que fue históricamente utilizado para distinguir ciertos síndromes heterogéneos, no caracterizados, del SUH-TS, ya que el término carece de especificidad y de sugerencia de causa. Asimismo, no se utiliza el término "idiopático" para ninguno de los síndromes MAT primarios.
La presencia de una anomalía causal, como la deficiencia de ADAMTS13 o una mutación del complemento, puede no expresarse clínicamente hasta que una condición, como el embarazo, una cirugía, o un trastorno inflamatorio, precipita un episodio agudo de MAT. El tratamiento de estos pacientes se centra en la causa del síndrome MAT primario, no en la condición precipitante. Estos pacientes son distintos de muchos otros pacientes que tienen anemia hemolítica microangiopática y trombocitopenia como manifestaciones de un trastorno subyacente (Tabla 2). El tratamiento de estos pacientes se centra en el trastorno subyacente.
El conocimiento de la patogenia, el manejo y los resultados de los síndromes MAT primarios se ha acelerado en los últimos años. El objetivo de esta revisión es proporcionar una perspectiva unificada de estos síndromes.
Tabla 1. Síndromes de Microangiopatía Trombótica (MAT) Primarios*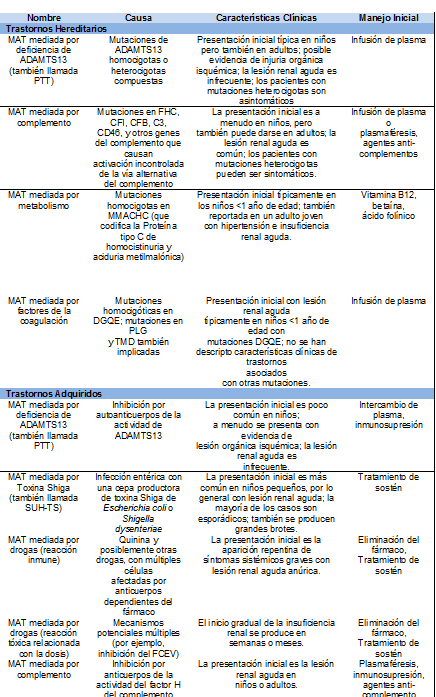
* Los síndromes MAT principales se describen mediante pruebas que apoyan una causa definida. La MAT mediada por Toxina Shiga (también llamada Síndrome Urémico Hemolítico relacionado con toxina Shiga [SUH-TS]) se presenta principalmente en niños y puede ser el más común de los nueve síndromes MAT primarios. Entre los adultos, la púrpura trombocitopénica trombótica (PTT) adquirida puede ser el síndrome MAT primario más común; la PTT adquirida es rara en niños, en los que la incidencia puede ser similar a la de PTT hereditaria. Se desconocen las frecuencias de las MAT que están mediadas por el complemento, el metabolismo, la coagulación, o drogas. La demostración de anticuerpos que pueden neutralizar la actividad del factor H del complemento sugiere que puede ocurrir MAT adquirida mediada por una deficiencia en el factor H del complemento. DGQE denota Diacilglicerol Quinasa ε, PLG Plasminógeno, TMD trombomodulina, y FCEV factor de crecimiento endotelial vascular.
| Tabla 2. Trastornos comunes asociados con anemia hemolítica microangiopática y trombocitopenia.* |
| • Infección sistémica • Cáncer sistémico • Preeclampsia severa, eclampsia, síndrome HELLP • Hipertensión severa • Trastornos autoinmunes (por ejemplo, lupus eritematoso sistémico, esclerosis sistémica, síndrome antifosfolipídico) • Trasplante de órganos o de células madre hematopoyéticas |
1. PTT (Adquirida y Hereditaria)
Antecedentes
En 1924, Moschcowitz describió a una niña de 16 años de edad con debilidad, palidez, púrpura, y hemiparesia quien murió después de 14 días por insuficiencia cardíaca. La autopsia reveló trombos hialinos en las arteriolas y los capilares terminales a lo largo de la mayoría de los órganos, incluyendo los riñones. Este informe fue la primera descripción de una MAT, presumiblemente una PTT, también llamada MAT mediada por deficiencia de ADAMTS13.
Causa
En 1982, se observaron multímeros de factor de von Willebrand inusualmente grandes en pacientes con PTT (hereditaria) crónica, en recaída. Este hallazgo condujo al descubrimiento de una proteasa de escisión del factor de von Willebrand, que se caracterizó posteriormente como ADAMTS13. La ADAMTS13 cliva multímeros del factor von Willebrand que son secretados desde las células endoteliales vasculares. La deficiencia de ADAMTS13 resulta en multímeros del factor de von Willebrand inusualmente grandes, con riesgo de trombos plaquetarios en vasos de pequeño calibre.
La PTT hereditaria (también llamada síndrome de Upshaw-Schulman) es causada por mutaciones homocigotas o heterocigotas compuestas de la ADAMTS13. Los pacientes con mutaciones heterocigotas no tienen anormalidades aparentes. La PTT adquirida es una enfermedad autoinmune causada por la inhibición por autoanticuerpos de la actividad de la ADAMTS13. La incidencia de PTT adquirida es mucho mayor en adultos (2,9 casos por 1 millón por año) que en niños (0,1 casos por 1 millón por año). Los factores que están asociados con una mayor frecuencia de este trastorno incluyen la edad entre 18 y 50 años, la raza negra, y el sexo femenino.
Cuadro clínico y diagnóstico Entre los síndromes MAT principales, la PTT es única porque rara vez causa daño renal agudo severo. Las características clínicas de la PTT hereditaria son episodios recurrentes de anemia hemolítica microangiopática y trombocitopenia, a menudo con alteraciones neurológicas u otros signos de lesión orgánica. El diagnóstico de PTT hereditaria requiere la documentación de la deficiencia de ADAMTS13 y la ausencia de un autoanticuerpo inhibidor de la misma, y la confirmación requiere la documentación de mutaciones de la ADAMTS13.
La PTT hereditaria puede ser evidente al nacimiento, con anemia hemolítica microangiopática y trombocitopenia, o no ser evidente hasta la edad adulta, cuando puede ser precipitada por una condición como el embarazo. Aunque la severidad de la condición puede estar relacionada con mutaciones de ADAMTS13, las observaciones de heterogeneidad entre hermanos sugieren que las manifestaciones clínicas requieren de factores genéticos o ambientales adicionales, similares a las observaciones en ratones deficientes de ADAMTS13.
Las características clínicas presentes en la PTT adquirida son diversas; algunos pacientes tienen alteraciones mínimas, mientras que otros están críticamente enfermos. Son comunes la debilidad, los síntomas gastrointestinales, la púrpura, y las alteraciones neurológicas focales transitorias. Sin embargo, un tercio de los pacientes no tienen anormalidades neurológicas. La mayoría de los pacientes tienen valores de creatinina normales o ligeramente elevados sólo de manera transitoria.
Los criterios de diagnóstico son la presencia de anemia hemolítica microangiopática y de trombocitopenia sin otra causa aparente. Por lo tanto, a veces no es posible la exclusión de otros síndromes MAT primarios. Un nivel de ADAMTS13 que indica menos del 10% de actividad normal apoya el diagnóstico clínico de PTT adquirida. Identifica casi todos los pacientes con riesgo de recaída, pero este nivel no es ni suficientemente sensible para identificar a todos los pacientes con PTT ni suficientemente específico para excluir a los pacientes con trastornos subyacentes.
Tratamiento
El tratamiento para la PTT hereditaria es el reemplazo de la ADAMTS13 mediante infusión de plasma. Los pacientes con reacciones alérgicas graves al plasma han sido efectivamente tratados con concentrado de factor VIII derivado de plasma que contiene ADAMTS13. Aunque muchos pacientes requieren plasma sólo cuando se producen trombocitopenia o síntomas, otros pueden requerir infusiones de plasma profilácticas en forma regular.
Antes del uso de la plasmaféresis, la sobrevida de los pacientes con PTT adquirida era del 10%. En 1991, un estudio aleatorizado controlado documentó una tasa de sobrevida del 78% con la plasmaféresis. La alta mortalidad sin tratamiento crea la urgencia de empezar el intercambio de plasma, que a menudo lleva a tratar a pacientes que no tienen PTT. Los glucocorticoides son el tratamiento estándar; el rituximab y otros agentes inmunosupresores son apropiados cuando el curso clínico es complicado. Raramente se requiere diálisis.
Resultados a largo plazo
Los resultados a largo plazo de los pacientes con PTT hereditaria son desconocidos. Los datos experimentales sugieren que la ADAMTS13 proporciona protección contra la aterosclerosis, pero no se sabe si los pacientes con PTT hereditaria tienen un mayor riesgo de enfermedad cardiovascular. El seguimiento a largo plazo de los pacientes con PTT adquirida puso de manifiesto el riesgo de recaída y un aumento de la prevalencia de deterioro cognitivo, depresión mayor, lupus eritematoso sistémico, hipertensión y muerte.
Necesidades futuras
Si el seguimiento a largo plazo muestra que la PTT hereditaria causa un aumento de la morbilidad, el tratamiento profiláctico será el punto más importante. El desarrollo de ADAMTS13 recombinante haría que el tratamiento profiláctico sea más simple y más seguro. Para el tratamiento de pacientes con PTT adquirida, se requieren alternativas más seguras y más accesibles que la plasmaféresis.
2. MAT mediada por Complemento (Adquirida y Hereditaria)
Antecedentes
Esta MAT que se caracteriza predominantemente por falla renal y se describe como SUH fue reconocida como un trastorno familiar en 1975. En 1981, se halló que dos hermanos con MAT tenían una deficiencia del factor H del complemento. La asociación entre la MAT y mutaciones en el gen que codifica el factor H del complemento (FHC) se estableció en 1998. Posteriormente, se identificaron mutaciones en múltiples otros factores que facilitan el aumento de la activación de la vía alternativa del complemento en pacientes con MAT.
Causa
La MAT mediada por complemento resulta de la activación incontrolada de la vía alternativa del complemento. A diferencia de las otras dos vías de activación del complemento, la vía alternativa es constitutivamente activa como resultado de la hidrólisis espontánea de C3 a C3b.
En ausencia de una regulación normal, la deposición de C3b sobre los tejidos puede aumentar notablemente, lo que resulta en aumento de la formación del complejo terminal C5b-9 del complemento (también llamado complejo de ataque de membrana) y lesión de las células normales. El papel preciso de la desregulación del complemento en la MAT no ha sido plenamente definido. Puede estar involucrada la lesión endotelial así como la desregulación del complemento en la superficie de las plaquetas causando activación.
La MAT mediada por complemento hereditaria puede resultar de una mutación con pérdida de función de un gen regulador (FHC, CFI, o CD46) o de una mutación con ganancia de función en un gen efector (CFB o C3). La mayoría de las mutaciones del complemento que se asocian con MAT son heterocigotas, incluso aunque muchos miembros de la familia con mutaciones heterocigotas sean asintomáticos.
Puede explicar esta discrepancia una diferencia entre probandos y miembros de la familia en la presencia de genes modificadores adicionales. Fueron identificadas otras anormalidades genéticas en pacientes con MAT mediada por complemento, incluyendo polimorfismos de un único nucleótido en FHC y CD46, variaciones del número de copias en los genes 1 y 3 relacionados con el FHC (FHCR1 y FHCR3), y fusión de genes de la región FHCR con FHC causado por recombinación homóloga no alélica. Estas anomalías genéticas adicionales pueden contribuir a la pérdida de regulación de la vía alternativa y al aumento del riesgo de MAT.
Además de las anomalías genéticas, una deficiencia funcional en el factor H del complemento puede resultar de anticuerpos para el complemento, dando lugar a una MAT adquirida. Estos anticuerpos FHC se corresponden con aproximadamente el 10% de las MAT mediadas por complemento. Estos anticuerpos son responsables de la protección celular defectuosa dependiente del FHC.
Cuadro clínico y diagnóstico
La lesión renal aguda y la hipertensión son alteraciones prominentes en la MAT mediada por complemento. Los criterios diagnósticos actuales son aquellos que se utilizaron en ensayos clínicos con un total de 37 pacientes, y que apoyaron la aprobación de eculizumab (un anticuerpo monoclonal humanizado que bloquea la generación de C5a y C5b) para el tratamiento del "SUH atípico" en 2011.
Estos criterios incluyen todo lo siguiente: un nivel de creatinina sérica en o por encima del límite superior del rango normal, anemia hemolítica microangiopática, trombocitopenia, actividad de ADAMTS13 del 5% o más, y análisis de materia fecal negativo para infección por germen productor de toxina Shiga.
Estos criterios no son específicos; también pueden ocurrir en todos los otros síndromes MAT primarios, así como en otros pacientes con anemia hemolítica microangiopática y trombocitopenia. Los estudios genéticos del complemento, ahora disponibles en el mercado con resultados rápidos, pueden proporcionar un diagnóstico más específico. Los niveles plasmáticos normales de C3, C4, y factores de complemento H, B, e I no excluyen el diagnóstico de MAT mediada por complemento.
Tratamiento
Puede utilizarse terapia anti-complemento para complementar el tratamiento con plasma y adelantarse a un potencial trasplante hepático. El eculizumab es actualmente el único agente anti-complemento disponible. Su efecto puede ser limitado en los pacientes que tienen mutaciones de C5. Los criterios diagnósticos no específicos y el hecho de que los pacientes con mutaciones del complemento no identificadas pueden tener una respuesta a la terapia anti-complemento hacen que sea difícil tomar la decisión de utilizar un agente anti-complemento como terapia inicial.
La terapia anti-complemento es un tratamiento inicial razonable para los pacientes con anticuerpos contra el factor H del complemento. Sin embargo, debe considerarse el uso de la inmunosupresión para reducir el título de anticuerpos. El alto costo del eculizumab (costo de adquisición al por mayor de 1 año de tratamiento para un adulto, $ 614,736; Farmacia del Centro Médico de la Universidad de Oklahoma, 5 de marzo de 2014) y que debe continuarse indefinidamente son temas críticos. Debe considerarse el riesgo de infección meningocóccica en relación con el tratamiento con eculizumab.
Resultados a largo plazo
Antes del uso de eculizumab, los riesgos de insuficiencia renal en fase terminal o muerte y de recurrencia después de un trasplante de riñón eran principalmente dependientes del análisis mutacional, con las mutaciones del FHC causando el mayor riesgo. Se supone que el tratamiento anti-complemento mejorará los resultados.
Necesidades futuras Son necesarios ensayos clínicos para determinar la duración más adecuada de la terapia anti-complemento y para desarrollar marcadores de vigilancia para confirmar la remisión renal. Se requiere más investigación genética para explicar la heterocigosidad y la baja penetrancia de las mutaciones del complemento actualmente identificadas y también para determinar la causa de enfermedad en pacientes que pueden tener MAT mediada por complemento, pero en quienes no se ha identificado una mutación.
3. Síndrome Urémico Hemolítico mediado por Toxina Shiga
Antecedentes
El nombre "Síndrome Urémico Hemolítico" se propuso por primera vez en 1955. Las características clínicas típicas del SUH-TS fueron descriptas por primera vez en un reporte de 1962 que implicó a cinco niños de 6 a 10 meses de edad que habían tenido diarrea previa a la falla renal. Se sospechó una causa infecciosa, pero la asociación entre SUH-TS e infección entérica, Shigella dysenteriae tipo 1, no se reconoció hasta 1975. No fue sino hasta 1983, durante una investigación de brotes de colitis hemorrágica, que la Escherichia coli O157:H7 fue identificada como un patógeno.
El mismo año, se describió la asociación entre SUH-TS y E. coli productora de toxina Shiga. Múltiples cepas de E. coli producen toxina Shiga; la E. coli O157:H7 es el patógeno más común asociado con SUH-TS en Europa y América. S. dysenteriae tipo 1 sigue siendo una causa endémica de SUH-TS en otros países. Aunque el SUH-TS se popularizó por sus grandes brotes, la mayoría de las ocurrencias son esporádicas. El SUH-TS es mucho más común entre los niños (edad media, 2 años), en los cuales la mortalidad es del 3%. El SUH-TS en adultos es más grave, con mayor mortalidad.
Causa
E. coli productora de toxina Shiga es una bacteria intestinal común en el ganado, en consonancia con el predominio rural del SUH-TS endémico. Los brotes provienen de productos contaminados, como agua, carne vacuna, verduras y otros alimentos. El daño celular se produce cuando la toxina Shiga se une a la globotriaosilceramida (Gb3, también conocida como CD77 o trihexósido de ceramida) de las células endoteliales, así como a las células mesangiales y epiteliales renales (podocitos y células tubulares).
La apoptosis celular resulta de la unión a Gb3, endocitosis, transporte retrógrado, y translocación citosólica de la toxina Shiga, con la subsiguiente inactivación ribosomal. La toxina Shiga también es activadora celular, pro-inflamatoria, y pro-trombótica y facilita la trombosis mediante la inducción de la secreción de factor von Willebrand.
Cuadro clínico y diagnóstico
Se manifiesta por dolor abdominal intenso y diarrea, a menudo sanguinolenta, varios días después de consumir alimentos contaminados. La trombocitopenia y el fallo renal comienzan con la resolución de los síntomas gastrointestinales. La toxina Shiga se identifica mediante análisis de materia fecal durante la fase de diarrea aguda, pero puede no ser identificable cuando comienza el SUH-TS.
Tratamiento
El tratamiento sigue siendo de sostén. La hidratación temprana, agresiva, tiene un rol de protección renal. Los pacientes comúnmente requieren diálisis. Los beneficios de la plasmaféresis y del tratamiento anti-complemento son inciertos.
Resultados a largo plazo
La hipertensión y las anomalías neurológicas pueden persistir después de que resuelva la fase aguda. Rara vez ocurre enfermedad renal en fase terminal.
Necesidades futuras
La mayor necesidad es la prevención de las infecciones enterohemorrágicas y del SUH-TS mediante medidas de salud pública (por ejemplo, seguridad e higiene de los alimentos). Nuevos agentes que neutralizan la toxina Shiga pueden proporcionar un tratamiento eficaz para el SUH-TS.
4. MAT mediada por Drogas (Reacción Inmune)
Antecedentes
Las reacciones adversas a drogas pueden ser causadas por reacciones inmunológicas, idiosincrásicas, no relacionadas con la dosis o por efectos tóxicos que son dependientes de la dosis y el tiempo. Ambos tipos de reacciones se asocian con MAT. La reacción inmune en la MAT mediada por fármacos fue reconocida por primera vez en 1980 en un paciente que había repetido episodios de insuficiencia renal aguda, hemólisis y trombocitopenia después de exposiciones documentadas a quinina.
Estudios posteriores confirmaron el patrón de recurrencia e implicaron más a la quinina al describir anticuerpos dependientes de quinina que eran reactivos con múltiples tipos celulares. Aunque se asociaron muchas otras drogas con MAT, sólo la MAT asociada a quinina ha sido apoyada por la documentación de anticuerpos fármaco-dependientes. Quetiapina y gemcitabina son las otras drogas para las que la asociación con episodios agudos de MAT ha sido apoyada por episodios agudos recurrentes ante exposiciones repetidas.
Causa
La unión del anticuerpo fármaco-dependiente a los antígenos de múltiples células puede ser facilitada por elementos estructurales que contienen los fármacos y que son complementarios a los dominios tanto del epítope como del anticuerpo. Los anticuerpos dependientes de quinina pueden mediar la MAT, en parte, por la activación de células endoteliales.
Cuadro clínico y diagnóstico
Se puede sospechar MAT mediada por drogas ante la aparición repentina de síntomas sistémicos graves, a menudo con lesión renal aguda anúrica, en cuestión de horas después de la exposición al fármaco. Puede haber un historial de enfermedad tras exposiciones previas al fármaco sospechoso. La asociación con la quinina puede ser pasada por alto porque la exposición puede ocurrir a lo largo de los años y puede no ser reportada por el paciente sin preguntas explícitas. La exposición puede incluir comprimidos o bebidas que contienen quinina. La documentación de anticuerpos fármaco-dependientes apoya el diagnóstico clínico; sin embargo, una prueba negativa no excluye la asociación con drogas.
Tratamiento
El tratamiento de sostén y la evitación del fármaco pueden ser las únicas medidas beneficiosas. Es frecuente que se inicie la plasmaféresis por la sospecha de PTT y porque la causa mediada por fármacos es incierta.
Resultados a largo plazo
Es frecuente la enfermedad renal crónica con hipertensión. Puede ocurrir enfermedad renal en etapa terminal.
Necesidades Futuras
Se requieren pruebas para la detección de anticuerpos fármaco-dependientes rápidamente disponibles para ayudar en el diagnóstico clínico. Entender el mecanismo de lesión renal aguda puede dar una idea para el tratamiento objetivo.
5. MAT mediada por Drogas (Reacción Tóxica Relacionada con la Dosis)
Antecedentes
Muchos fármacos, incluyendo agentes inmunosupresores y quimioterapéuticos e inhibidores del factor de crecimiento endotelial vascular (FCEV), fueron reportados como causa de MAT mediante toxicidad dosis y tiempo dependiente. La evidencia que apoya una relación causal es limitada.
Causa Puede haber múltiples mecanismos de lesión renal mediada por drogas tóxicas. Entre los probables roles de los inhibidores de la calcineurina (como la ciclosporina y el tacrolimus) está su capacidad de causar disfunción endotelial y aumento de la agregación plaquetaria, posiblemente a través de la inhibición de la prostaciclina. La inhibición de la función del FCEV en las células endoteliales y los podocitos renales causa el desarrollo gradual de MAT glomerular.
Cuadro clínico y diagnóstico
La presentación típica es la pérdida gradual de la función renal con hipertensión. Se puede producir MAT abrupta, severa, al igual que con un mal uso intravenoso del opiáceo oximorfona.
Tratamiento
El tratamiento de apoyo y la evitación del fármaco pueden ser el único manejo beneficioso. Para algunos medicamentos, tales como los inhibidores de la calcineurina, la reducción de la dosis, más que la evitación del fármaco, puede ser suficiente.
Resultados a largo plazo
La anemia hemolítica microangiopática y la trombocitopenia suelen resolver. La insuficiencia renal puede persistir.
Necesidades futuras
Es esencial determinar los fármacos que provocan MAT, o identificar explicaciones alternativas para la MAT en estos pacientes.
6. MAT mediada por Metabolismo
Antecedentes
La Enfermedad de la Cobalamina C es un trastorno hereditario del metabolismo de la cobalamina (vitamina B12) que puede causar MAT y disfunción orgánica múltiple en los lactantes. Además, se ha reportado MAT en un adulto.
Causa
Los trastornos del metabolismo de la cobalamina resultan de mutaciones homocigotas o heterocigotas compuestas en un gen que codifica la proteína tipo C de la aciduria metilmalónica y la homocistinuria (MMACHC). La deficiencia resultante de metilcobalamina causa hiperhomocisteinemia, disminución de los niveles plasmáticos de metionina, y aciduria metilmalónica. El metabolismo anormal de la cobalamina C se asocia con activación plaquetaria, generación de especies de oxígeno reactivo, disfunción endotelial, expresión aumentada del factor tisular, y activación de la coagulación.
Cuadro clínico y diagnóstico
Los lactantes con enfermedad de la cobalamina C tienen diversas anomalías del desarrollo. El único adulto reportado se presentó con anemia hemolítica microangiopática, trombocitopenia, insuficiencia renal aguda, e hipertensión; no tenía antecedentes personales o familiares relevantes.
Tratamiento
La hidroxicobalamina parenteral es el principal tratamiento para los niños. Cuando el paciente adulto antes mencionado no tuvo una respuesta al tratamiento anti-complemento, se sospechó un defecto intracelular del metabolismo de la vitamina B12 y se confirmó mediante pruebas de rutina disponibles, que mostraron hiperhomocisteinemia, disminución de metionina en plasma, aciduria metilmalónica, y niveles plasmáticos normales de vitamina B12. El paciente respondió al tratamiento con hidroxicobalamina, betaína, y ácido folínico.
Resultados a largo plazo
Las secuelas neurológicas son comunes en los lactantes afectados. Se reportaron enfermedad renal crónica con hipertensión y proteinuria hasta en un 40% de los pacientes. La enfermedad renal en etapa terminal y la recurrencia de MAT parecen prevenirse con el tratamiento de reemplazo.
Necesidades futuras
Se requiere mayor conciencia de la MAT inducida por cobalamina C y un mayor uso de las pruebas de detección de rutina para la deficiencia de cobalamina.
7. MAT mediada por Factores de la Coagulación
Antecedentes
Se identificaron anomalías genéticas de la trombomodulina, el plasminógeno, y una proteína asociada a la proteína quinasa C, diacilglicerol quinasa ε (DGQE), en pacientes con MAT. Estos hallazgos sugieren que puede haber un papel primordial para las proteínas de la coagulación en la patogénesis de los Síndromes MAT.
Causa
La cuestión de si el papel de la trombomodulina en la MAT está principalmente relacionado con la coagulación o si está mediada únicamente por el complemento requiere mayor estudio. Se documentó el rol de la DGQE en dos informes que describen 22 pacientes en 12 familias. Todos los pacientes tenían mutaciones homocigotas o heterocigotas compuestas de la DGQE. Los pacientes con mutaciones heterocigotas no tenían anomalías clínicas. En un informe no se identificaron mutaciones de genes reguladores del complemento.
Se propuso que una mutación homocigota en el gen que codifica la DGQE conduce a una pérdida de función, resultando en activación de la proteína quinasa C (PQC). La activación de la PQC facilita la sobre regulación de factores protrombóticos (factor de von Willebrand, factor tisular, e inhibidor del activador del plasminógeno tipo 1) y la baja regulación del receptor del FCEV.
La disminución de la función del FCEV con la consiguiente lesión de podocitos renales también puede ser una consecuencia de mutaciones de la DGQE. La suma de estos eventos es un cambio hacia un estado protrombótico. La comunicación entre el sistema de la coagulación y el sistema del complemento puede tener un papel en la patogénesis de la MAT.
Cuadro clínico y diagnóstico
Los pacientes con mutaciones DGQE se presentan con lesión renal aguda. La mayoría de estos pacientes estudiados eran menores de 1 año.
Tratamiento Los beneficios de la infusión o el intercambio de plasma y la inmunosupresión fueron inconsistentes. Hubo recaídas mientras los pacientes estaban recibiendo terapia anti-complemento.
Resultados a largo plazo
Es común la enfermedad renal en etapa terminal. Cuatro niños con este trastorno fueron sometidos a trasplante de riñón; uno tuvo injuria renal recurrente.
Necesidades futuras
Es esencial la determinación de la prevalencia de estos síndromes y la comprensión de los mecanismos que conducen a la MAT.
Mecanismos de lesión orgánica
Las causas antes mencionadas de síndromes MAT primarios no explican por qué la lesión renal es predominante en todos salvo en la PTT. La PTT causa trombos en la extensa microvasculatura renal, aunque la lesión renal aguda severa y la enfermedad renal crónica son raras. La diferencia puede ser que la patogénesis de la PTT se relaciona principalmente con trombosis vascular, mientras que la patogénesis de los otros síndromes MAT primarios también incluye daño a las células renales y células endoteliales residentes.
Por ejemplo, en pacientes con SUH-TS, la toxina Shiga causa daño a los podocitos epiteliales, células mesangiales, y células tubulares renales. Los inhibidores del FCEV afectan a los podocitos epiteliales glomerulares, y la mayor activación de la proteína quinasa C en pacientes con mutaciones de la DGQE puede causar injuria en los podocitos. Comprender el mecanismo de lesión en células renales residentes en la MAT mediada por complemento, por anticuerpos quinina-dependientes, o en la Enfermedad de la Cobalamina C requiere más estudio.
También hay claras diferencias con respecto al riesgo de enfermedad renal crónica entre los siete síndromes MAT primarios en los que la lesión renal aguda está normalmente presente. Entre los síndromes adquiridos, rara vez se desarrolla lesión renal crónica en pacientes con SUH-TS, tal vez porque el SUH-TS no recurre.
Sin embargo, los pacientes con un solo episodio reconocido de MAT inducido por quinina comúnmente tienen lesión renal crónica. Entre los síndromes hereditarios, también hay una variedad sustancial en la gravedad de la lesión renal.
Los pacientes con mutaciones del complemento pueden presentar lesión renal aguda a cualquier edad, mientras que aquellos con mutaciones DGQE suelen presentar lesión renal aguda en la infancia. Estas variaciones en las manifestaciones clínicas de los síndromes MAT enfatizan la necesidad de una mayor comprensión de los mecanismos de la enfermedad.
Conclusiones
En los últimos años, ha habido una aceleración dramática en la comprensión de los síndromes MAT primarios. Los nuevos descubrimientos con respecto a los mecanismos causales crearon oportunidades para tratamientos específicos. La disponibilidad de tratamientos específicos llevó a la necesidad de diagnósticos rápidos y precisos. El uso de tratamientos eficaces ha disminuido la mortalidad, pero también ha puesto de manifiesto morbilidades a largo plazo anteriormente no reconocidas. Estos avances predicen la aceleración continua de la comprensión de los principales síndromes MAT.
Comentario: Los síndromes de microangiopatía trombótica son un conjunto heterogéneo de trastornos hereditarios o adquiridos, cuyas características clínicas incluyen anemia hemolítica microangiopática, trombocitopenia, y daño orgánico variable. La presencia de una anomalía causal puede no expresarse clínicamente hasta que cierta condición (como embarazo, cirugía, o proceso inflamatorio) precipita el episodio agudo de MAT, y el tratamiento se centra básicamente en la misma.
Si bien han aumentado notablemente los conocimientos con respecto a los síndromes MAT primarios, se requieren nuevas investigaciones que ayuden a la comprensión de estos trastornos, a fin de mejorar las opciones de tratamiento y disminuir la morbi-mortalidad que surge de los mismos.
Resumen y comentario objetivo: Dra. María Eugenia Noguerol
Suscribirse a:
Entradas (Atom)